
Remember me






To establish a highly efficient protein extraction method from a sample of human hair, we evaluated six different methods, as shown in Fig. 1A. The six methods compared overall protein extraction efficiency using three different hair fragmentation techniques and three different protein extraction methods. SDS-PAGE gels stained with Coomassie blue showed that the GH and LN-BH methods did not yield visible proteins (Fig. 1B and C). The RTO and LN-H95 methods yielded moderate amounts of protein with average extraction efficiency. However, these two methods were unstable, as only two of the three biological replicates showed visible protein. The SH-65 method extracted proteins from hair with higher efficiency and stability, as evident from highly consistent SDS-PAGE banding patterns across all three biological replicates. However, we observed that proteins with molecular weights under 40 kDa were almost entirely absent. The H-90-SH-65 method yielded the largest amounts of protein from hair, with highly reproducible, evenly distributed protein across all three experimental replicates (Fig. 1B). Quantitative results based on the stained SDS-PAGE gel showed that the average efficiency of the H-90-SH-65 method for extraction is 22.71% higher than the SH-65 method, and compared with other extraction methods, the efficiency is increased by at least 1.3 times (Fig. 1C). These results demonstrated that both strong shock and boiling can effectively improve the efficiency of protein extraction from hair.
Fig. 1
Development of a highly efficient protein extraction method from hair. A Workflow for the development of a highly efficient protein extraction method from hair. B Silver stained SDS-PAGE to compare the protein extraction efficiency among six methods. GH: Glass Homogenizer; LN-BH: Ground with Liquid Nitrogen (LN) followed by homogenize with Silica beads (BH); RTO: RT Oscillation; LN-H95: Ground with Liquid Nitrogen followed by Heating at 95℃; SH-65: Shaking at 65℃; H-90-SH-65: Heating at 90℃ for 12 h followed by Shaking at 65℃ for 12 h. C Quantitative analysis of the silver stained SDS-PAGE of B.* indicates p < 0.05. *** indicates p < 0.001. **** indicates p < 0.0001
Development of high efficiency protein extraction buffer from hairTo investigate the impact of buffer components on the hair protein extraction efficiency, we compared the buffer solution containing a high concentration of SDS (lysis buffer 1) and high concentration of urea (lysis buffer 2) with the highest efficiency H-90-SH-65 extraction method (Fig. 2A). The gel electrophoresis results showed that SDS buffer produced more uniformly dispersed protein bands throughout the lane of the Coomassie brilliant blue stained gel compared to Urea buffer (Fig. 2B). The stained SDS-PAGE quantitative analysis revealed that protein extracted using the SDS buffer had 1.67 times higher yield than that of the Urea buffer (Fig. 2C). The effective extraction of proteins from hair can be achieved through combining long-term heating and strong impact with a simple high-concentration SDS lysis buffer. In comparing hair sample processing methods and optimizing buffer components, we determined that the combination of H-90-SH-65 and SDS-containing protein lysis solution demonstrated the highest extraction efficiency, and hence, we termed this technique the PLEE (PTM Lab for protein extraction from hair with high efficiency) method.
Fig. 2
Development of high efficiency protein lysis buffer. A Workflow for the development of highly efficient protein lysis buffer from hair. B Coomassie brilliant blue stained SDS-PAGE to compare the proteins extracted with two lysis buffers. C Quantitative analysis of Coomassie blue stained SDS-PAGE of B
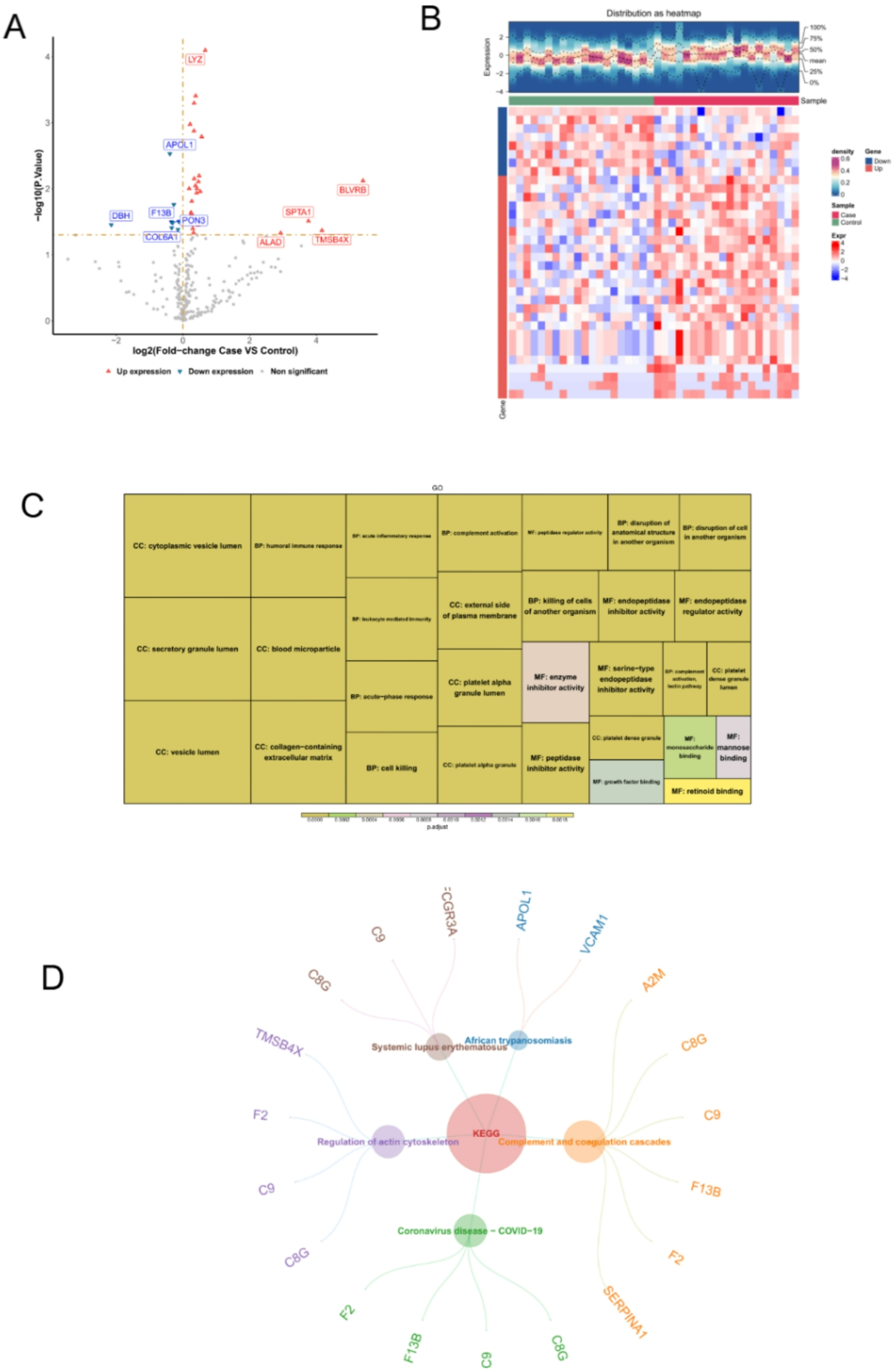
Comparison of protein extraction efficiency between PLEE method and Mase methodTo validate the protein extraction efficiency of the PLEE method for hair, we compared the efficiency of protein extraction of the PLEE method with the method reported by Mase laboratory (Mase method). To evaluate the efficiency of the two methods, we used normal hair and damaged hair for protein extraction (Fig. 3A). Equal amounts of protein were separated by 10% SDS-PAGE (Fig. 3B). The results showed that the PLEE method extracted significantly more hair protein than the Mase method, with a PLEE method protein extraction efficiency being 3.30 times that of the Mase method in both normal hair and damaged groups (Fig. 3C). Using consistent liquid phase and mass spectrometry conditions for both methods, peptide segment samples were prepared from an equivalent amount of protein, and proteomics data was obtained. The total number of identified proteins, keratin, and keratin-associated proteins in the two methods were compared. The results showed that the PLEE method identified 175 proteins, including 52 keratin-associated proteins and 51 keratin, while the Mase method identified 155 proteins, including 51 keratin-associated proteins and 50 keratin, respectively. Therefore, the PLEE method had a better total number of identified proteins (as indicated in Figs. 2, 3, 4 and 5 C-D), consistent with the SDS-PAGE observation. Using consistent liquid phase and mass spectrometry conditions for both methods, peptide segment samples were prepared from an equivalent amount of protein, and proteomics data was obtained. The total number of identified proteins, keratin, and keratin-associated proteins in the two methods were compared. The results showed that the PLEE method identified 175 proteins, including 52 keratin-associated proteins and 51 keratin, while the Mase method identified 155 proteins, including 51 keratin-associated proteins and 50 keratin, respectively. Therefore, the PLEE method had a better total number of identified proteins (as indicated in Figs. 2, 3, 4 and 5C-D), consistent with the SDS-PAGE observation (Fig. 3D-E).
Fig. 3
Comparison of protein extraction amount between two methods. A workflow comparing the efficiency of the PLEE method with the Mase method for the extraction of hair proteins. B Coomassie brilliant blue stained SDS-PAGE to compare the quantity and quality of proteins extracted by the two methods. C Quantitative analysis of Coomassie blue stained SDS-PAGE of B. D Venn diagram of the total number of protein identification by the two methods. E Venn diagram of the total number of KRT and KAP identification by the two methods. * indicates p < 0.05; KRT: keratin; KAP: keratin-associated proteins
Fig. 4
High-quality large-scale proteome based on the PLEE method characterizes hair injury by bleaching damage. A Venn diagram of 80.6% protein in the overlap of normal and damaged samples. B Venn diagram of 92.3% keratin and keratin-associated proteins in the overlap of normal and damaged. C The correlation among biological replicates and different conditions. D Cluster heatmap for damaged and normal groups; E. PCA analysis for damaged and normal groups. F Volcano plots showing the fold changes for damaged and normal data. The cut off were folded change > 2 and p-value < 0.05. G GO and KEGG term analysis of down-regulated proteins
Fig. 5
The resolution of which high-quality large-scale proteomics data used Mase method to distinguish samples is insufficient. A Venn diagram of 73.5% protein in the overlap of normal and damaged. B Venn diagram of 84.3% keratin and keratin-associated proteins in the overlap of normal and damaged. C The correlation among biological replicates and different conditions. D Cluster heatmap for damaged and normal groups. E. PCA analysis for damaged and normal groups. F Volcano plots showing the fold changes for damaged and normal data. The cut off were folded change > 2 and p-value < 0.05
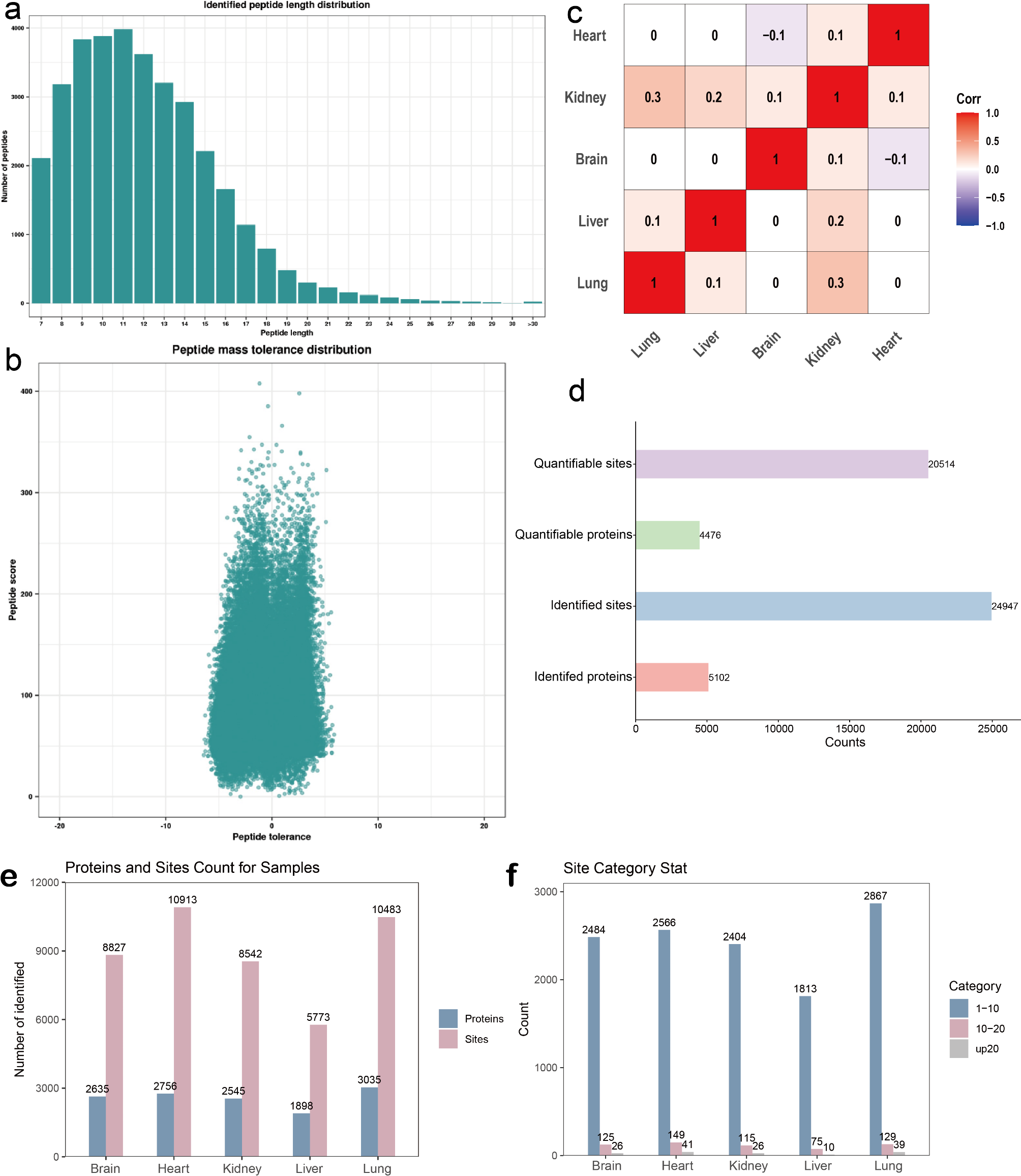
High-quality large-scale proteome based on the PLEE method.We identified 155–158 proteins in both normal and damaged hair samples, of which 48–51 were keratin and keratin-associated proteins. Between the samples, 80.6% of total proteins and 92.3% of keratin and keratin-associated proteins overlapped (Fig. 4A-B). On average, 6 peptides were identified per protein (Table 1). The number of identified hair proteins was slightly different between normal and damaged hair samples. Figure 4C shows a high correlation between technical replicates, with a Pearson correlation of 0.96–0.97. In contrast, the correlation between different treatment groups was 0.91–0.94, which is lower than the correlations observed in both technical replicates.
Table 1 Results of proteomics database searchUsing principal component analysis and cluster analysis, we were able to distinguish between normal and damaged hair samples, which can be separated into two distinct groups (Fig. 4D-E). On the volcano plot in Fig. 4F, we filtered differentially expressed proteins between the two groups using a twofold change and p-value less than 0.05. We identified 16 down-regulated proteins, including 11 keratin and keratin-associated proteins, and 14 up-regulated proteins. The analysis of differentially expressed proteins revealed that the down-regulated proteins were enriched in various processes related to hair structure, such as the hair cycle, aging, intermediate filaments, keratinization, and others, which may contribute to the deterioration of hair quality caused by damage (Fig. 4G).
The resolution of which high-quality large-scale proteomics data used Mase methodIn normal and damaged hair samples, we identified 134–142 proteins, of which 48–52 keratin and keratin-associated proteins and 734–790 peptides. Between samples, 73.5% of the total protein and 84.3% of the keratin and keratin-associated proteins between the samples were overlapped (Fig. 5A-B). On average, 5 peptides were identified per protein (Table 2). The number of identified hair proteins between normal and damaged hairs was changed slightly. As shown in Fig. 5C, there is a high correlation between technique replicates, with Pearson correlation between 0.94–0.95. The correlation between different treatment groups was 0.91–0.95, which is lower than those in both technical replicates.
Table 2 Results of proteomics database searchThe principal component analysis (PCA) and cluster analysis suggested that normal and damaged hair samples could not be clearly distinguished (Fig. 5D-E), and the entire sample set could be divided into two groups. We used a volcano plot to identify differentially expressed proteins between the two groups with a twofold change and p-value less than 0.05, resulting in the identification of 2 down-regulated keratin-associated proteins (KRTAP1-5 and KRTAP1-2) and 7 up-regulated proteins (as shown in Fig. 5F). Overall, our results indicate that the Mase method is inferior to the PLEE method in terms of the depth of protein identification and insufficient at distinguishing between samples.
The protein sequence coverage of PLEE method was greater than that Mase methodWe compared the two hair protein extraction methods by analyzing the distribution of protein sequence coverage. Firstly, two methods jointly identified 26 keratins (Fig. 6A). The sequence coverage of 14 keratins by the PLEE method was greater than that by the Mase method (Fig. 6B). Second, t two methods jointly identified 19 keratin-associated proteins (Fig. 6C). The sequence coverage of 11 keratin-associated proteins in the PLEE method was greater than that in the Mase method (Fig. 6D). These results further supported that the PLEE method was more efficient than the Mase method.We compared the protein sequence coverage of normal and damaged hair samples using both PLEE and Mase methods. For PLEE, out of the 26 keratins and 26 keratin-associated proteins identified, the sequence coverage of 15 keratins and 16 keratin-associated proteins in the normal group was greater than that in the damaged group. We further plotted the 10 proteins with the highest difference and observed that the distribution of the normal group was more concentrated than that of the damaged group. Similarly, for Mase, out of the 32 keratins and 19 keratin-associated proteins identified, the sequence coverage of 9 keratins and 9 keratin-associated proteins in the normal group was greater than that in the damaged group (Fig. 7A-E). We also plotted the 10 proteins with the highest difference and observed a more concentrated distribution in the normal group than in the damaged group (Fig. 7F).
Fig. 6
The sequence coverage of the identified proteins by the PLEE method wa s greater than that by the Mase method. A Donut chart of keratin identification by the two methods. B Boxplots of sequence coverage of keratin-associated proteins. C Donut chart of keratin-associated proteins identification by the two methods. D Boxplots of sequence coverage of keratin
Fig. 7
The sequence coverage of the identified proteins in the normal group was higher than that in the damaged group. A Boxplots of sequence coverage of keratin-associated proteins for the PLEE method. B Boxplots of sequence coverage of keratin for the PLEE method. C Scatter plot of sequence coverage distribution of TOP10 proteins. for the PLEE method. D Boxplots of sequence coverage of keratin-associated proteins for the Mase method. E Boxplots of sequence coverage of keratin for the Mase method. F Scatter plot of sequence coverage distribution of TOP10 proteins. for the Mase method
Comments (0)