Data source
The RNA sequencing and clinical data (survival time and survival status) of patients with GC and other tumors were collected from the Genotype Tissue Expression Project (GTEx) database and the Cancer Genome Atlas (TCGA) database.
Pathological sample collection
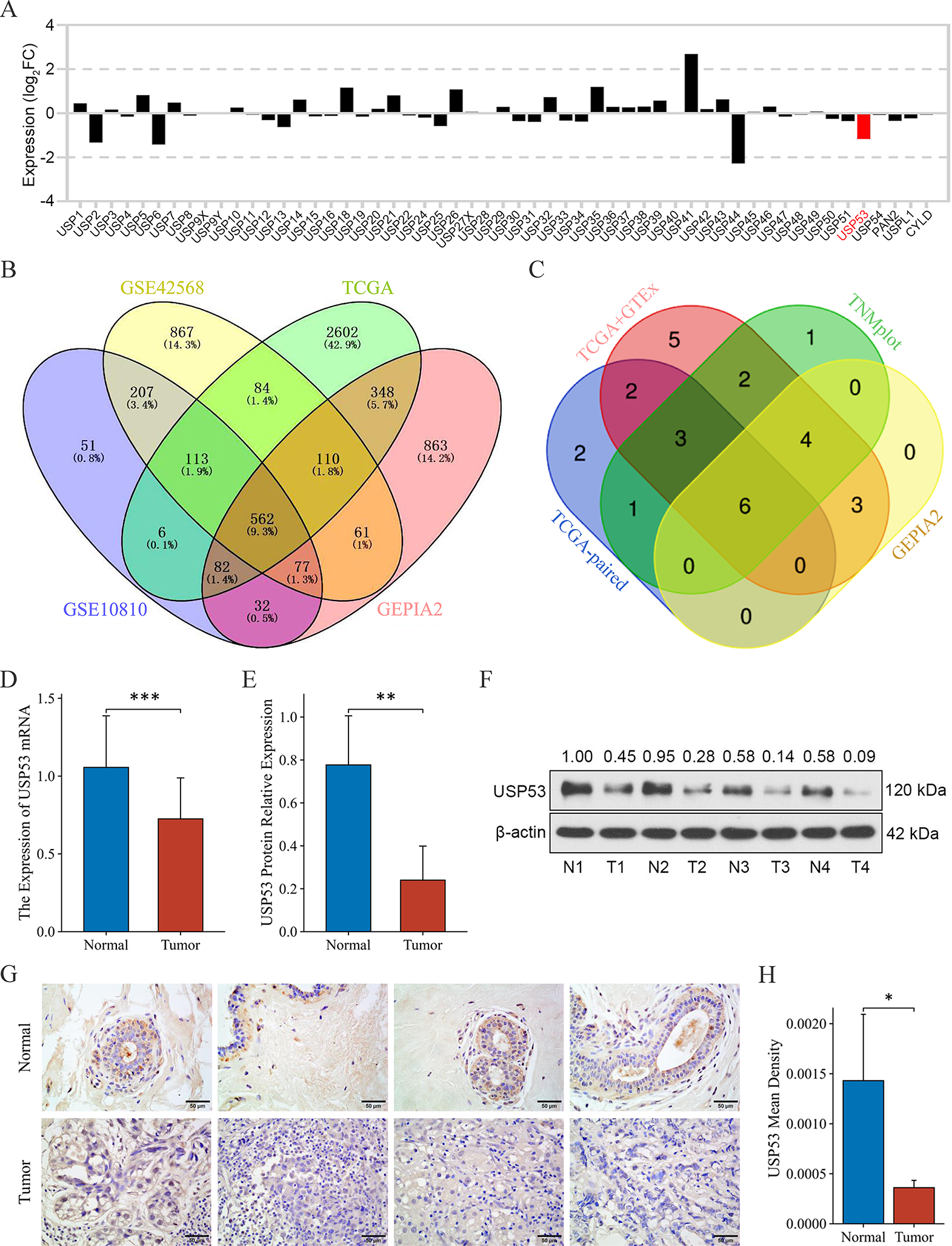
A total of 3 samples of gastric cancer tissues and their matched paracancerous tissues were collected between March 2023 and April 2023 at the Affiliated Hospital of Jiangsu University. This study was approved by the medical ethics committees of the Affiliated Hospital of Jiangsu University and was conducted in line with the Declaration of Helsinki.
Immunohistochemistry (IHC)
Tumor tissues and paracancerous tissues were fixed in 10% formalin, paraffin-embedded, sliced into 4 ~ 6 μm sections, and placed onto slides. After deparaffinization, rehydration and microwave antigen retrieval, the slides were incubated with MBD3 (Proteintech, Cat No. 14258–1-AP) antibody at a 1:800 dilution at 4 °C overnight. Then, the slides were incubated with secondary antibody at room temperature for 30 min and stained with DAB substrate, followed by hematoxylin counterstaining.
Cell culture
The human gastric normal epithelial cell line GES-1 and GC cell lines (SGC-7901, MGC-803 and HGC-27) were stored by the Institute of Medical Science, Jiangsu University (Zhenjiang, Jiangsu, China). All cells were tested and authenticated by short tandem repeat analysis. Cells were cultured in DMEM (Meilunbio, Dalian, China) containing 10% fetal bovine serum, 1% penicillin and streptomycin (HyClone, South Logan, UT, USA) in a humidified incubator at 37 °C and 5% CO2.
Western blotting
Cells were rinsed with cold PBS, and then lysed with RIPA buffer containing 1% PMSF, 1% protease inhibitor, 5% 2-mercaptoethanol and 93% 2 × loading buffer. The cell lysate boiled at 100 °C for 10 min was centrifuged at 12 000 × g for 10 min. Subsequently, the total protein was separated by SDS-PAGE and transferred onto PVDF membranes for immunoblot assays. Membranes were blocked with 5% BSA for 1 h at room temperature and incubated with primary antibodies overnight at 4 °C. The membranes were washed, incubated with the respective HRP-conjugated secondary antibody for 1 h at room temperature, and then visualized with an ECL detection system (Amersham Pharmacia Biotech, Little Chalfont, UK). The antibodies were: rabbit anti-MBD3 (Proteintech, Cat No.14258–1-AP), rabbit anti-β-Tubulin (Abcam, CAT 21058), rabbit anti-MMP2 (ImmunoWay, CAT YT2798), rabbit anti-MMP9 (ImmunoWay, CAT YT1892), rabbit anti-Flag (ABclonal, CAT AE063), rabbit anti-Snail (Cell Signaling, CAT 3879), rabbit anti-α-SMA (Cell Signaling, CAT 68463), rabbit anti-Vimentin (Cell Signaling, CAT 5741), rabbit anti-N-cadherin (Cell Signaling, CAT 13116), rabbit anti-β-catenin (Cell Signaling, CAT 8480), rabbit anti-E-cadherin (Cell Signaling, CAT 3195), rabbit anti-p-PI3K (Sigma, CAT SAB4504314), rabbit anti-PI3K (Santa Cruz, CAT sc-1637), rabbit anti-p-AKT (Santa Cruz, CAT sc-271966), rabbit anti-AKT (Santa Cruz, CAT sc-5298), rabbit anti-p-mTOR (Santa Cruz, CAT sc-293133), rabbit anti-mTOR (Santa Cruz, CAT sc-517464), rabbit anti-ACTG1 (Affinity, CAT AF0115).
Quantitative real-time polymerase chain reaction (qRT-PCR)
RNAiso Plus (Invitrogen) was used to extract total RNA according to the manufacturer’s protocol. Complementary DNAs (cDNAs) were synthesized from RNA samples (1 μg) by RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific). qRT-PCR was performed via a SYBR Green Mix kit (Bio Rad Laboratories, Hercules, CA). Analysis of the relative expression was based on the 2−ΔΔCt method. The primers were as follows: GAPDH-forward: 5'-GGTGAAGGTCGGTGTGAACG-3' and GAPDH-reverse: 5'-CTCGCTCCTGGAAGATGGTG-3'; MBD3-forward: 5'-CGGCCACAGGGATGTCTTTT-3' and MBD3-reverse: 5'-TGCTGGGGTGGTTGGTAATC-3'; MMP2-forward: 5'-CACAGGAGG AGAAGGCTGTG-3' and MMP2-reverse: 5'-GAGCTTGGGAAAGCCAGGAT-3'; MMP9-forward: 5'-TTCAGGGAGACGCCCATTTC-3' and MMP9-reverse: 5'-TGTAGAGTCTCTCGCTGGGG-3'.
Prognostic analysis
Kaplan–Meier plots were used to assess the relationship between MBD3 expression and the prognosis (OS, Overall survival, DSS, Disease-specific survival) of cancers. The area under the curve (AUC) curves were generated by analyzing the data using the timeROC package and the results were visualized by ggplot2. Risk score maps were visualized with the ggplot2 package. The survival package was used for proportional hazards hypothesis testing and Cox regression analysis. The rms package was used for calibration analysis and visualization. The survival package was used for proportional hazards hypothesis testing and Cox regression analysis, and the rms package was used to construct and visualize the nomogram correlation model. The forest map was visualized by ggplot2. The proportional hazards hypothesis test and fitted survival regression were performed using the survival package, and the results were visualized using the survminer package and the ggplot2 package. Hypothesis testing was performed using the log-rank test, and P < 0.05 was considered statistically significant.
Cell transfection
The plasmids sh-EGFP, sh-MBD3, 3 × Flag-vector (shown as Vector) and Flag-MBD3 were obtained from the Institute of Medical Science, Jiangsu University (Zhenjiang, Jiangsu, China). The sequence was confirmed through DNA sequencing. Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) was used for cell transfection based on the manufacturer’s protocols. Each well of a 6-well plate contained 2 μg plasmids and 10 μL Lipofectamine 2000.
Cell migration and invasion assay
Transwell assays were performed in a Transwell chamber (pore diameter, 8 μm; MilliporeSigma). Transfected GC cells were resuspended in serum-free medium, and subsequently seeded into the upper layer of the chamber (6 × 104 cells/100 μl) while complete medium was added to the chamber bottom. After 20 h, the cells on the lower surface were fixed with 4% polyformaldehyde (Aladdin, Shanghai, China) and stained with 0.05% crystal violet (Beyotime, Shanghai, China) for 30 min at room temperature whereas the cells on the upper surface were wiped slightly. An inverted light microscope (Olympus Corporation) was used to capture the images. The cell invasion assay was similar to the migration assay except that cells were seeded in Matrigel-coated Transwell inserts (BD Bioscience, Corning, NY, USA).
Wound healing assay
Transfected GC cells (2 × 105/well) were seeded in 24-well plates for 24 h and then scratched in wells with a 10 μl micropipette tip. Nonadherent cells were washed away with phosphate-buffered saline (PBS), and fresh medium was added. Images of wounds were acquired at 0 h and 20 h with a microscope. The wound-healing rate was calculated as follows: 100% × [(wound width at 0 h − width at 20 h)/width at 0 h].
Cell Counting Kit (CCK)-8 assay
After 48 h of transfection, 1 × 103 cells/100 μL were seeded into 96-well plates in each well. After 12 h, the cells adhered to the wall. Then, the cells were incubated with 100 μl CCK8 reagent mixture (10 μl CCK8 reagent: 90 μl DMEM) without light for 2 h at 37 °C. The results were measured at 450 nm absorbance by a microplate reader (Bio-Rad, Hercules, CA, USA) and analyzed via GraphPad Prism version 8.
Colony formation assay
Single-cell suspensions were plated in 6-well plates at 1 × 103 cells/well and maintained at 37 °C and 5% CO2 for 10 ~ 14 days. The supernatant was changed every three days. Finally, 4% paraformaldehyde was used to fix colonies and 0.5% crystal violet was used for staining for 30 min. The number of visible colonies was counted by ImageJ.
Xenograft mouse model
The protocol was approved by the Ethics Committee of Jiangsu University. MGC-803 cells (2 × 106 cells/site) stably transfected with vector or Flag-MBD3 were subcutaneously injected into 5-week-old BALB/c nude mice (Shanghai SLAC Laboratory Animal Co., Ltd., Shanghai, China) to generate xenografts. There were five female mice in each group. Tumor volume was measured weekly after injection and calculated using the formula: length × width × height × π/6 (the tumor is ellipsoid).
RNA sequencing
MGC-823 cells (3 × 105) were seeded into a 6-well culture plate and transfected with plasmids for 48 h. Cells were washed with cold PBS twice, and total RNA was isolated using RNAiso Plus (Takara) following the manufacturer's protocol. After verifying its concentration and integrity, the qualified RNA samples were subjected to PCR amplification to construct a cDNA library. Cluster generation and sequencing were performed on a NovaSeq 6000 S4 platform using a NovaSeq 6000 S4 Reagent kit V1.5. To guarantee the data quality that was used for analysis, the useful Perl script was used to filter the original data to remove low-quality sequences. The reference genomes and the annotation file were downloaded from the ENSEMBL database (http://www.ensembl.org/index.html).
Statistical analysis
Each experiment was performed separately at least three times. The data are presented as the mean ± standard deviation (SD). Student’s t-test determined the statistical significance between two groups, while two-way analysis of variance (ANOVA) was used to analyze data from more than 3 groups. The results were analyzed via GraphPad Prism 8 software. P < 0.05 was considered statistically significant.



Comments (0)