
Remember me
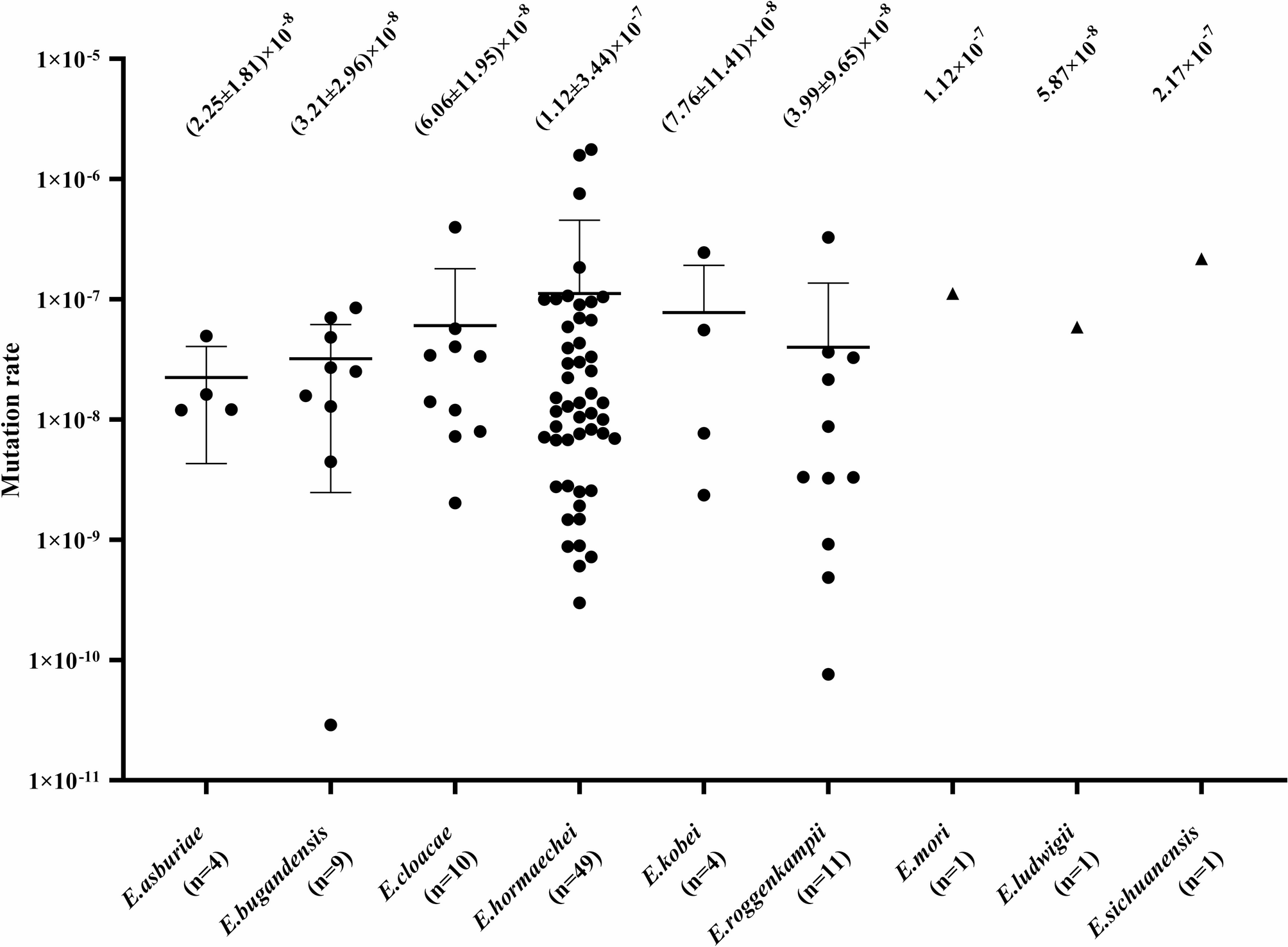





While being housed in our aquaria, a small number of sea urchins contracted spotting disease, which allowed us to observe disease progression in a controlled environment. Diseased sea urchins showed one or two discrete regions of blackened necrotic tissue of varying sizes (Fig. 1). For all infected sea urchins, progression of the infection was similar. Initially the lesions were small and nearly undetectable; however, as the infection continued, the epidermal tissue was degraded, resulting in the loss of all appendages within the lesioned area (Fig. 1). When the lesions expanded and deepened, the underlying test was exposed, and sea urchins lost the primary spines in areas surrounding the lesion. Eventually, the test disintegrated, which signaled that a sea urchin would succumb soon afterwards, indicating that spotting disease was fatal. The duration of infection typically lasted months to years, however, the final phase of infection after the test was degraded tended to develop rather quickly as a sea urchin became moribund. In this late stage of the disease, sea urchins demonstrated altered behaviors, which, in addition to primary spine loss in non-lesioned regions, included cessation of eating and failing to hold on to anything with their tube feet (i.e., aquarium wall, kelp, holding box). In all cases, infected sea urchins never showed signs of recovery from spotting disease. Spotting disease was not communicable, as illustrated when sea urchins with lesions were housed in the same aquarium with healthy sea urchins, and the disease never appeared on the healthy animals.
Fig. 1
Sea urchins with spotting disease show discrete lesions at the equatorial to ventral body regions. All sea urchins are positioned with the oral surface facing down. A Diseased sea urchin (D1) has a single large black necrotic lesion. This sea urchin displays unusual orientation of its spines, which point in various directions rather than uniformly perpendicular to the body surface. This is a behavioral indication of disease that has been noted previously [15]. B Diseased sea urchin (D2) has two large black necrotic lesions, labelled “a” and “b”. Parts of the test (white) are exposed around the outer region of lesion a (arrows). This sea urchin has lost all primary spines, including non-lesioned areas of the body surface, which indicates its moribund condition. C Diseased sea urchin (D3) has one large black necrotic lesion. This sea urchin shows typical spine orientation of perpendicular to the body surface, which indicates better health despite the lesion. Exposed test is also evident within the lesion (arrow). D Diseased sea urchin (D4) has a small black necrotic lesion of approximately 1 cm in diameter. The mouth is located on the ventral side (yellow arrow). This sea urchin also shows indications of better health, including primary spines generally pointing perpendicular to the body surface
The four infected sea urchins were sampled at different stages of spotting disease as inferred from the different sizes and numbers of lesions (Fig. 1). Diseased animal 1 (D1) had one large lesion and at the time of sacrifice, it was not eating consistently, which was an indication of impending mortality (Fig. 1A). Diseased animal 2 (D2) had two lesions of different sizes that were referred to as ‘a’ and ‘b’ for the larger and smaller lesions, respectively (Fig. 1B). At the time of sacrifice, D2 had lost all primary spines, was no longer feeding, and did not display tube foot function to attach to the box in which it was housed, all of which were indicators of its moribund state. Diseased animal 3 (D3) had one large lesion but exhibited normal tube foot function, all spines were present except for those surrounding the lesion, and it was eating normally (Fig. 1C). Sea urchins D1, D2, and D3 were all treated twice with pen/strep; once at half concentration, and a second time at the standard concentration, one week apart. The pen/strep treatments had no effect on resolving the discrete lesions. Diseased animal 4 (D4) had one very small lesion that was first noticed once it was approximately 1 cm in diameter (Fig. 1D). D4 did not show any other signs of altered behaviors associated with spotting disease. Unfortunately, this animal died due to unrelated factors and therefore was not used in the analysis of dissected tissues that were collected from the other sea urchins upon sacrifice. However, D4 samples were collected for the analysis of the global surface microbiome and LS.
Sequence data identifies ASVs in all samplesPreliminary analyses by PCR of the microbial community gDNA isolated from the samples of both diseased and healthy sea urchins confirmed that bacterial gDNA was present in in all samples. 16S rRNA gene sequencing resulted in a total of 81,536 non-chimeric reads (Table 2).
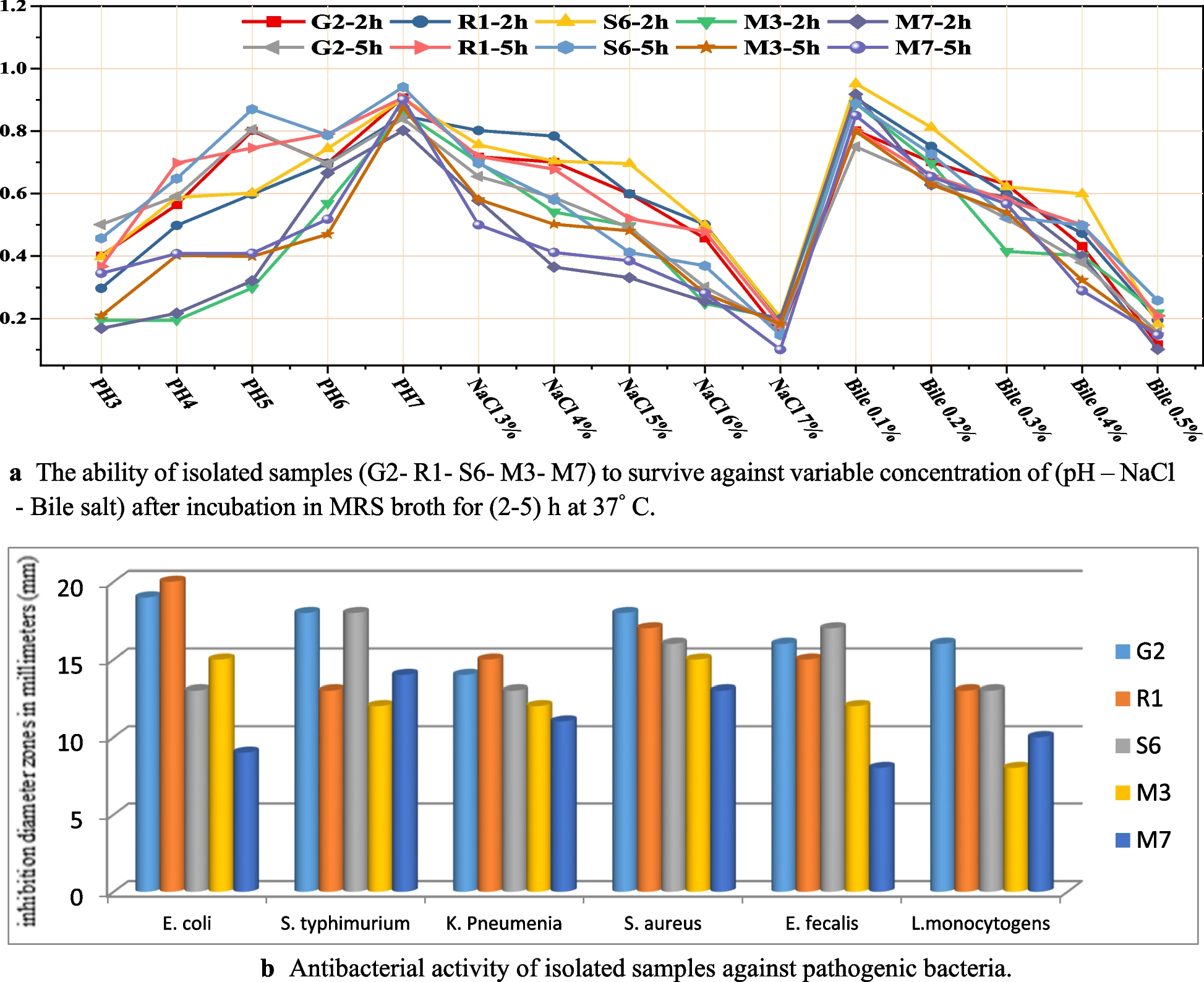
Table 2 Read processing summaryThe global surface microbiomes of diseased sea urchins show taxonomic differences compared to the global surface microbiomes of healthy sea urchinsThe global surface microbiomes on echinoids are poorly studied. However, an initial report shows that they are altered when variegated sea urchins, Lytechinus variegatus, are transferred from open water to a closed system [24]. The surface microbiomes also shift as sea urchins recover from BSUD, and it can differ on sea urchins housed in different aquaria [15]. Because there were healthy sea urchins in the same aquarium with spotting disease, and the disease was not communicable, we investigated the global surface microbiome to identify differences between diseased and healthy sea urchins. The alpha diversity of the global surface microbiomes on diseased and healthy sea urchins were analyzed using observed species, Chao1 [32], and ACE [33] (Fig. 2A-C). Results showed that there were no significant differences in alpha diversity between the global surface microbiomes on the diseased compared to the healthy sea urchins (ANOVA, p > 0.05), although the diseased group microbiome had decreased alpha diversity compared to the healthy group for all metrics. Beta diversity was analyzed by weighted UniFrac that measures microbiome composition, which showed overlap of the samples from diseased and healthy sea urchins (PERMANOVA, p > 0.05; Fig. 2D), indicating that the microbiome composition was not significantly different between the two groups.
Fig. 2
The global surface microbiome diversity is not different between diseased and healthy sea urchins. Alpha diversity is analyzed by A Observed Species, B Chao1, and C ACE. The box plots show the mean and quartile values for each group, which are not significantly different (ANOVA, p > 0.05). D Beta diversity is analyzed at the level of ASV sequences using weighted UniFrac and visualized with NMDS. Ellipses around sample groups show 95% confidence intervals assuming a multivariate t-distribution (solid line) or a multivariate normal distribution (dashed line). The beta diversity of the groups is not significantly different (PERMANOVA, p > 0.05), indicating that microbial composition is similar for the global surface microbiomes on diseased and healthy sea urchins housed in the same aquarium
The composition of bacterial taxa collected from the two groups of sea urchins were compared to identify differences among the global surface microbiomes, in addition to comparisons with the two samples of filtered aquarium seawater (fSW). The most abundant taxa were selected (Additional File; Tables S1, S2) to identify the similarities and differences. Although beta diversity showed no differences among the samples (Fig. 2D), the phyla present in the global surface microbiomes were different between the diseased and healthy sea urchins (Fig. 3). Samples were generally dominated by Proteobacteria, and three of the diseased surface microbiome samples had an elevated abundance of the phylum Bacteroidota compared to the healthy surface microbiome samples (Fig. 3A). There were also major differences in the genera identified in the global surface microbiomes among the groups (Fig. 3B). The microbial composition of the diseased surface microbiome samples differed from the healthy surface microbiome samples, and there were also differences among the samples within the diseased group. The samples collected from D1 and D2 had many taxa in common with elevated abundances of a genus in the Cryomorphaceae family, a genus in the Cyclobacteriaceae family, Lutibacter, and a genus in the Cellvibionaceae family. This was not evident in the global surface samples from D3 and D4 nor from the corresponding samples from the healthy sea urchins. The D3 and D4 surface samples were more similar to the healthy surface samples, and were mainly composed of Psychromonas, Colwellia, and a family in the Bacteroidia class. Samples from the fSW were distinct from all samples collected from the sea urchins. It is notable that sea urchins D3 and D4, with less extensive lesions, had global surface microbiomes that were more similar to those on the healthy sea urchins, whereas D1 and D2, with more extensive lesions, had microbiomes that were very different from the healthy sea urchins. LEfSe analysis showed that Cyclobateriaceae, Lutibacter and Pseudoteredinibacter were significantly differentially abundant in the diseased global surface microbiome samples, and that Gastranaerophilales and Enterobacterales were significantly differentially abundant in the corresponding samples from the healthy sea urchins (Fig. 4A; Additional File, Table S3). These results showed that the microbial composition of the global surface microbiome differed in samples from sea urchins that were in different stages of spotting disease progression. Results also showed that taxonomic differences were evident for the genera identified in the global surface microbiomes of the diseased compared to the healthy groups of the sea urchins that were housed in the same aquarium, and that they were also different from the microbes in the fSW samples. These results indicated that spotting disease could be characterized based on the differences in the global surface microbiomes of diseased compared to healthy sea urchins irrespective of the influences of the bacterial composition of the seawater in the aquarium and did not require direct sampling of the lesions.
Fig. 3
Global surface microbiomes of diseased vs. healthy sea urchins show taxonomic differences. A All identified phyla for the sea urchin surface samples and the fSW sample (Additional File, Table S1) illustrate the relative abundance of each taxon in each sample. B Genera with an average relative abundance of > 0.1% across all samples (Additional File, Table S2) are illustrated by the relative abundance per sample. fSW is the average of fSW1 and fSW2. Taxa in A and B that could not be assigned at the level of phylum or genus are listed as the most specific known taxonomic level. BD2-3 is in the order Victivallales, the Pir4 lineage is in the family Pirellulaceae, vadinHA49 is in the phylum Planctomycetota, JGI-0000069-P22 is in the class Gracilibacteria, MSBL3 is in the family Kiritimatiellaceae, and HOC36 is in the class Gammaproteobacteria. ASV sequences that could not be assigned to a phylum are grouped as Bacteria. Sample name abbreviations are defined in Table 1
Fig. 4
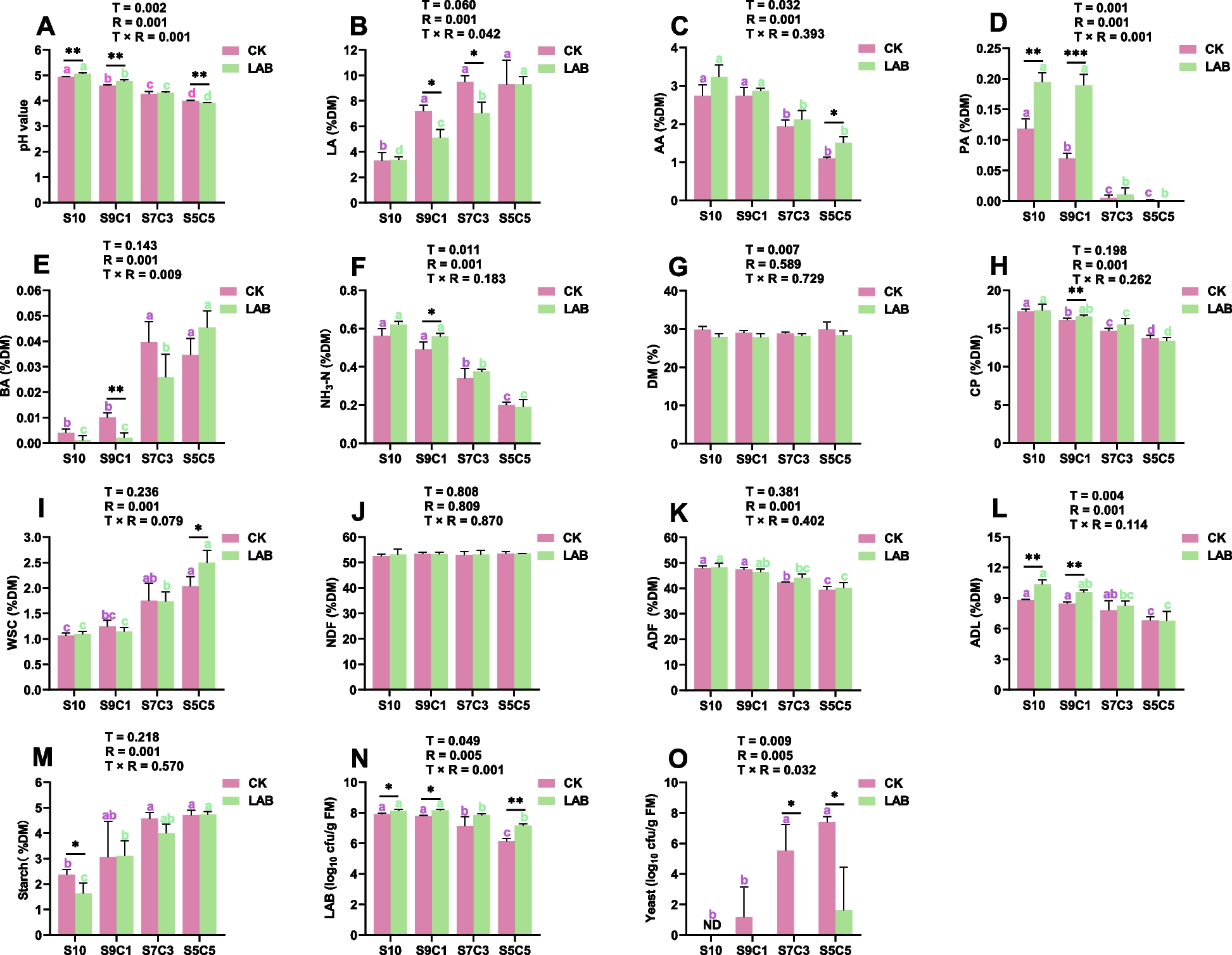
Many taxa are differentially abundant in the microbiome samples. A heatmap shows all taxa identified by LEfSe that are significantly differentially abundant (p < 0.05) and have an LDA score of > 2 as their abundance per group for A the global surface microbiome samples, B the lesioned body wall (LBW) and lesion surface (LS) microbiome samples, and C the tissue microbiome samples (Additional File, Tables S3, S6, S9). Sample name abbreviations are defined in Table 1
The lesions have a unique microbial compositionOne of the reported causative agents of spotting disease, Vibrio coralliilyticus, forms biofilms on the surface of tissues [4]. During the formation of a biofilm, bacteria aggregate, function as a single unit, and produce an extracellular matrix that protects the bacterial community from external factors such as antimicrobial agents [34]. Based on the appearance of the lesions on infected sea urchins and based on the lack of response to the pen/strep treatment by the diseased sea urchins, microbes associated with lesions may have formed a biofilm. To investigate this possibility, the LS was collected by the swab method to determine whether the microbial composition on the surface of the lesions differed from the microbes that were associated with the lesioned body wall tissues, as identified in the LBW group (Fig. 5). Results from Observed Species, Chao1, and ACE did not show significant differences between the microbiomes collected from LS compared to the LBW (Fig. 5A-C). Beta diversity showed that the LS samples clustered closely together compared to the LBW samples, which were less similar and more spread out with a large confidence interval (Fig. 5D). This indicated that the microbial composition among the LS samples was similar, and that there was more variation among the LBW samples. Furthermore, because the two clusters overlapped, the microbial compositions between the LS group and the LBW group likely had many shared taxa. The microbial composition in the sSW was very different from the samples collected from both the LS and the LBW. These results suggested that microbiome compositions were similar among the LS samples and less similar among the LBW samples, and were both different from the microbes in the aquarium seawater.
Fig. 5
The microbiome compositions are similar between the lesion surface and the lesioned body wall. Alpha diversity of the microbiomes from the LS and LBW sample groups are evaluated by A Observed Species, B Chao1, and C ACE. The box plots show the mean and quartile values for each group, which are not different (ANOVA, p > 0.05). D Beta diversity is evaluated at the ASV level using weighted UniFrac and visualized with NMDS. Ellipses around sample groups show 95% confidence intervals assuming a multivariate t-distribution (solid line) and a multivariate normal distribution (dashed line). The microbiomes of the LS compared to the LBW sample groups are not significantly different (PERMANOVA, p > 0.05). The sSW sample is shown for comparison. Sample name abbreviations are defined in Table 1
Further analysis of the microbiomes of the LS, LBW, and seawater samples (fSW and sSW) was carried out to compare the taxa identified in those sample groups (Fig. 6). At the level of phylum, the microbial composition of the LS and LBW microbiome samples were similar and were composed of mainly Proteobacteria and Bacteroidota, which varied inconsistently among the samples (Fig. 6A; Additional File, Table S4). At the level of genus, there were also many similarities in the microbial compositions (Fig. 6B; Additional File, Table S5), which were consistent with the beta diversity results (Fig. 5D). The LS samples from D1-D3 were highly similar to each other and were mainly dominated by a genus in the Cyclobacteriaceae family, a genus in the Cryomorphaceae family, and a genus in the Cellvibrionaceae family. The LS sample from D4, which had a single small lesion, differed from the others and was mainly composed of a genus in the Cellvibrionaceae family, the HOC36 group, and Pseudophaeobacter. The LBW samples also had an elevated abundance of a genus in the Cyclobacteriaceae family, and a genus in the Cryomorphaceae family, however, two of the LBW samples (D2a and D3) had elevated abundances of Vibrio and Candidatus Photodesmus. LEfSe results identified Cellvibrionaceae, Pseudophaeobacter, the HOC36 group, Gammaproteobacteria, and Roseobacter as significantly differentially abundant in the LS microbiome samples, whereas Canditatus Photodesmus was significantly differentially abundant in the LBW microbiome samples (Fig. 4B; Additional File, Table S6). Overall, all LBW samples were composed of a similar set of microbes.
Fig. 6
The microbiomes of the lesion surface and the lesioned body wall are highly similar. A All identified phyla are shown as the relative abundance in each sample (Additional File, Table S4). B Genera with an average relative abundance of > 0.1% across all groups (Additional File, Table S5) are illustrated by the relative abundance per sample. Abundance of taxa from replicated samples are averaged. Both types of seawater control samples are included for comparisons, which are the microbes collected from 500 ml of filtered seawater (fSW) and seawater collected with a swab (sSW). Taxa in A and B that could not be assigned at the level of phylum or genus are listed as the most specific known taxonomic level. Taxa that could not be assigned to a phylum are grouped under Bacteria. Sample name abbreviations are defined in Table 1. BD2-3 is in the order Victivallales, the Pir4 lineage is in the family Pirellulaceae, vadinHA49 is in the phylum Planctomycetota, JGI-0000069-P22 is in the class Gracilibacteria, and HOC36 is in the class Gammaproteobacteria
When the LS microbiome samples from the diseased sea urchins were compared to each other, as well as to the sSW sample, D1-D3 had similarly abundant taxa, however, the LS microbiome from D4 differed from the other LS microbiomes (Fig. 6). Although an initial aim was to compare the microbiomes of the LS samples with non-lesioned surface areas of sea urchins with spotting disease, attempts to collect microbial gDNA from the surfaces of non-lesioned tissue using the swab method was unsuccessful. The 16S rRNA gene could not be amplified from the DNA isolated from the non-lesion surface swab samples indicating that were likely too few microbes associated with surfaces of healthy sea urchins, in agreement with a previous report [7]. Although another, more destructive collection method has been reported with more success for isolating bacterial gDNA from sea urchin surfaces and spines [24], this approach would not have been comparable to the LS sampling that was collected with swabs. Consequently, comparisons to non-lesioned surface microbiomes collected by the swab method could not be carried out.
Comparisons of the abundant taxa in the microbial compositions of the seawater samples showed that sSW sample included a genus in the Cellvibrionaceae family, Microbacterium, and Psychromonas. The abundant taxa in the fSW samples consisted mainly of the HOC36 group, the Pir4 lineage, and Psychromonas. Differences in the microbial composition of these seawater samples collected from the same aquarium may be attributed to differences in the volume of seawater collected. The small water volumes collected by swabs for the sSW samples may not have acquired an evenly distributed sample of microbes. Nonetheless, when the LS microbiomes were compared to the seawater samples, there were many taxa in the LS microbiomes that were absent from the microbiomes collected from either of the aquarium seawater samples. Overall, results indicated that the LS microbiome was distinct from the microbes in the seawater, but that it was highly similar to the LBW microbiome, which suggested similar a microbial composition on the surface of the lesion and within the lesioned body wall.
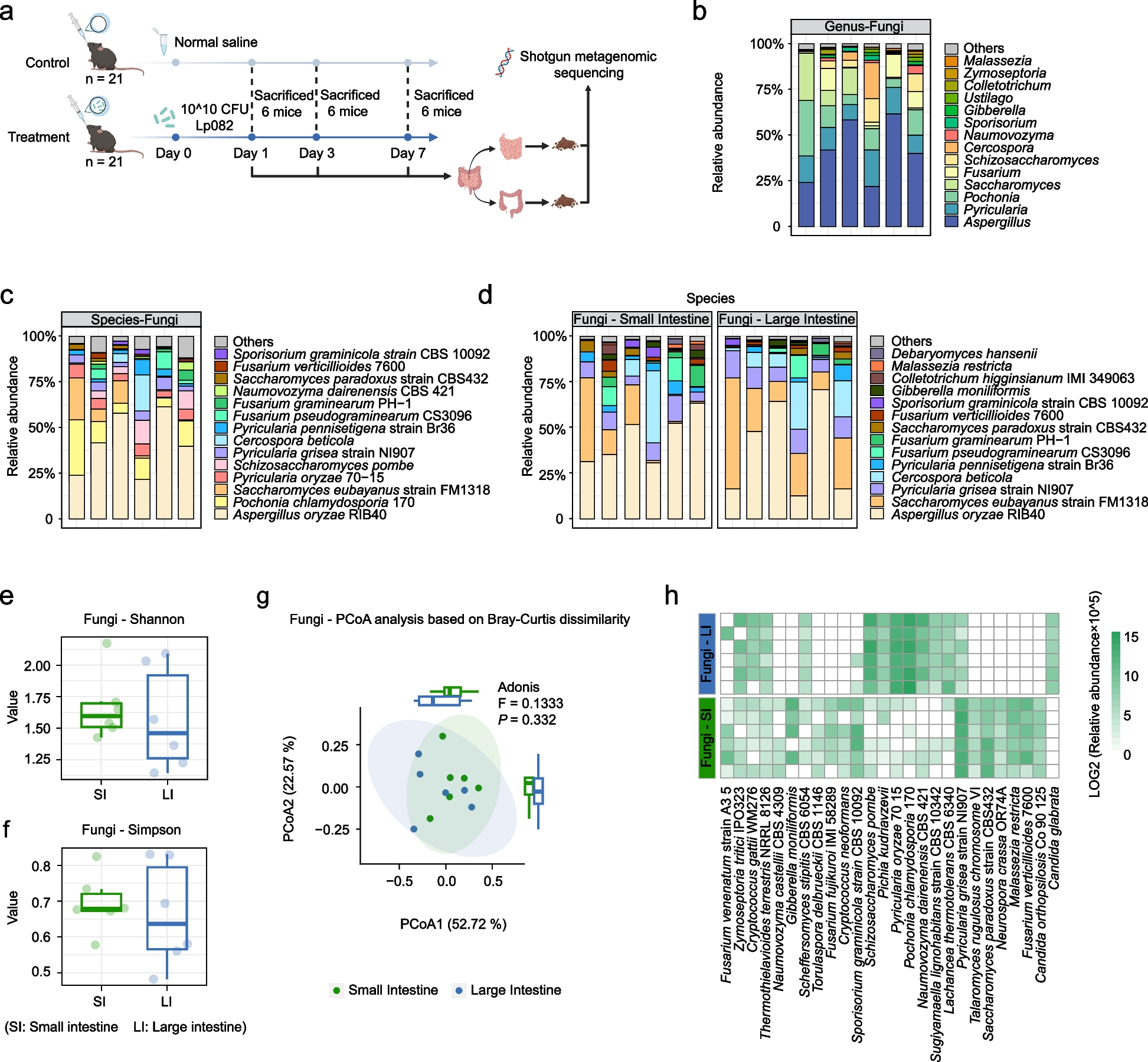
Microbiomes are different between diseased and healthy sea urchin tissuesA comparison of the microbiomes of dissected tissues from diseased compared to healthy sea urchins housed in the same closed aquarium system eliminates the variability of the microbial environment and is therefore informative for characterizing the microbiomes associated with the spotting disease infection on tissues that are outside of the lesioned areas. The alpha diversity metrics of the microbiomes from dissected tissues from infected sea urchins were evaluated and compared to each other and to the microbiomes of dissected tissues from healthy sea urchins (Fig. 7). Results from Observed Species, Chao1, and ACE, identified large variations in the samples within the groups of sea urchin tissues and therefore did not show significant differences among the groups (ANOVA, p > 0.05) (Fig. 7A-C). Beta diversity showed that the sample groups largely overlapped, but that the LBW samples tended to cluster together, and the HBW samples also clustered (Fig. 7D). The DBW samples had large variations, and therefore had a large confidence interval. The DCF samples tended to cluster with the LBW samples, whereas the HCF samples clustered with the HBW samples, indicating similar microbiome compositions for these pairs of sample groups. The beta diversity analysis revealed differing microbial compositions that suggested unique microbiomes for the LBW, DBW and HBW tissues.
Fig. 7
The microbiomes of dissected tissues from diseased and healthy sea urchins are different. Alpha diversity is analyzed by A Observed Species, B Chao1, and C ACE. The box plots show the mean and quartile values for each group, which are not significantly different (ANOVA, p > 0.05). D Beta diversity is analyzed at the ASV level using weighted UniFrac. Ellipses around sample groups show 95% confidence intervals assuming a multivariate t-distribution (solid line) and a multivariate normal distribution (dashed line). Groups are not significantly different (PERMANOVA, p > 0.05). Sample name abbreviations are defined in Table 1
To understand the taxa underlying the bacterial compositions of the dissected tissues, the phyla identified in the microbiomes were compared among the samples (Fig. 8). Differences in the microbial composition were evident among the different tissues based on the relative abundance of the phyla (Fig. 8A; Additional File, Table S7). All groups were dominated by Proteobacteria, however, the LBW group including D1, D2a, b, and D3 (Fig. 1), was also largely composed of Bacteroidia, which was generally only present in low abundances in the other samples. The DBW samples from D1, D2, and D3 were largely composed of Proteobacteria with a few other phyla of low abundance, which was similar to the DCF samples from the same animals. The HBW group and HCF group from the healthy sea urchins similarly had low abundances of Bacteroidia, as well as Actinobacteriota and Verrucomicrobia that were not detected in the LBW samples. The taxa were also compared at the level of genus to identify differences among the sample groups (Fig. 8B; Additional File, Table S8). All LBW samples had an elevated abundance of a genus of the Cryomorphaceae family and a genus of the Cyclobacteriaceae family. In addition, there was an elevated abundance of Candidatus Photodesmus and Vibrio in two of the four LBW samples. The DBW samples differed in their microbial compositions, which was consistent with the large variation shown by beta diversity. The DCF samples, similar to the DBW samples, had differing microbial compositions for different sea urchins. The HBW samples consisted of a few taxa in common, which included Psychromonas, a genus of the Cellvibrionaceae family, Colwellia, and Microbacterium, among others. The microbial composition of the HCF samples differed based on the sea urchin, however, there were many taxa that were also present in the HBW samples, which included BD2-3, Microbacterium, Psychromonas, and a family of the Bacteroidia class.
Fig. 8
The microbiome of each dissected tissue has a distinct microbial composition. A All phyla that were identified are shown as the relative abundance of each phylum in each sample (Additional File, Table S7). Taxa that could not be assigned to a phylum are grouped under Bacteria. B Genera with an average relative abundance of > 0.1% across all groups are shown as their relative abundance per sample (Additional File, Table S8). Taxa that could not be assigned at the level of genus are listed as the most specific known taxonomic level. Sample name abbreviations are defined in Table 1. BD2-3 is in the order Victivallales, vadinHA49 is in the phylum Planctomycetota, JGI-0000069-P22 is in the class Gracilibacteria, and MSBL3 is in the family Kiritimatiellaceae
Significant differential abundance of taxa for individual sample groups was identified by LEfSe analysis (Fig. 4C; Additional File, Table S9). Results showed that these taxa in the LBW samples were Candidatus Photodesmus, Cyclobacteriaceae, Cryomorphaceae, Vibrio, Pseudoteredinibacter, and Lutibacter. Taxa identified for the DCF samples were Pseudoalteromonas and Pseudophaeobacter, and taxa identified for the HCF samples were Microbacterium, BD2-3, Bacteroidia, vadinHA49, and Brevibacterium. Overall, the microbiome of the LBW was composed of a few taxa which were generally absent from the samples of the other groups. The microbiomes in the tissues from diseased sea urchins were distinct from those from healthy sea urchins, as evidenced by the differences between the DBW compared to the HBW samples, and the DCF compared to the HCF samples.
Spotting disease and BSUD may have different etiologiesDuring this study and as reported previously [15], we observed differences between spotting disease and BSUD that both occurred in our aquaria. After receiving a shipment of sea urchins and placing them in aquarium B, all animals from that shipment contracted BSUD (see Fig. 1 in [15]). In comparison, there were four sea urchins (D1-4) that were housed in aquarium C that originated from a different shipment and became infected with spotting disease. BSUD was communicable based on the spread of the disease to all animals in aquarium B, including a number of sea urchins that were transferred into aquarium B during the outbreak [15]. D3, with a single discrete spotting disease lesion, was one of the sea urchins transferred into aquarium B with animals infected with BSUD. It began to show symptoms of BSUD about 60 days after the transfer including agitated spine movement, tissue swelling at the base of the spines, and primary spine loss, consistent with the other sea urchins with BSUD [15]. As a result, sea urchin D3 appeared to be infected with both BSUD and spotting disease simultaneously and the symptoms of each disease could be discerned separately. D3 did not succumb to BSUD but followed the same infection and recovery process as the other animals in the aquarium, including primary spine regrowth over the course of 2–3 months. However, the spotting disease lesion on D3 did not resolve. BSUD and spotting disease appeared as separate diseases based on their different symptoms and different recoveries, particularly for D3.
Comments (0)