Challenging the Traditional Approach for Interpreting Genetic Variants: Lessons from Fabry Disease
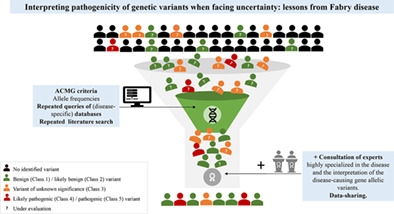
Fabry disease (FD) is an X-linked genetic disease due to pathogenic variants in GLA. The phenotype varies depending on the GLA variant, alpha-galactosidase residual activity, patient's age and gender and, for females, X chromosome inactivation. Over 1,000 variants have been identified, many through screening protocols more susceptible to disclose non-pathogenic variants or variants of unknown significance (VUS). This, together with the non-specificity of some FD symptoms, challenges physicians attempting to interpret GLA variants. The traditional way to interpreting pathogenicity is based on a combined approach using allele frequencies, genomic databases, global and disease-specific clinical databases, and in silico tools proposed by the American College of Medical Genetics and Genomics. Here, a panel of FD specialists convened to study how expertise may compare with the traditional approach. Several GLA VUS, highly controversial in the literature (p.Ser126Gly, p.Ala143Thr, p.Asp313Tyr), were re-analysed through reviews of patients' charts. The same was done for pathogenic GLA variants with some specificities. Our data suggest that input of geneticists and physicians with wide expertise in disease phenotypes, prevalence, inheritance, biomarkers, alleles frequencies, disease-specific databases, and literature greatly contribute to a more accurate interpretation of the pathogenicity of variants, bringing a significant additional value over the traditional approach.

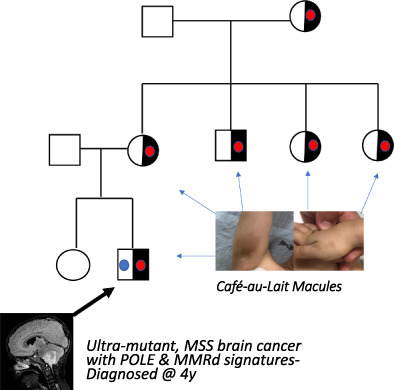
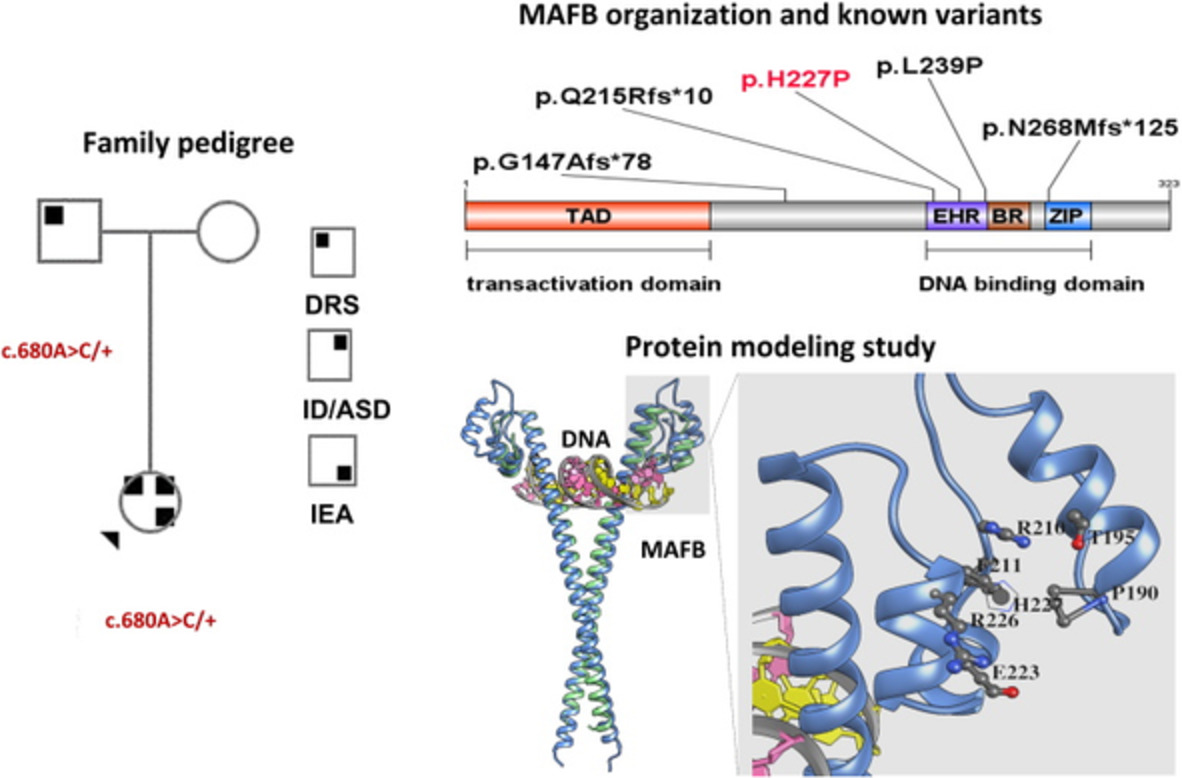
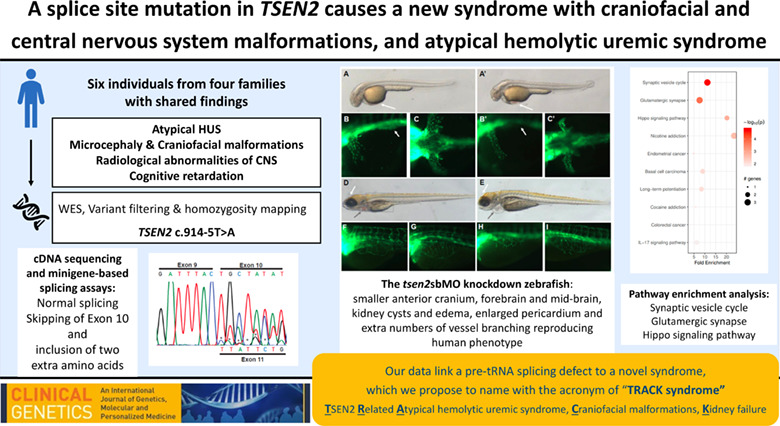

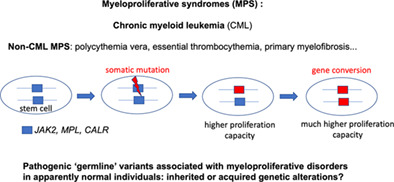

Comments (0)