

Remember me
 Table 1. Clinical Features of GNAS Mutation Carriers with Severe Obesity.
Table 1. Clinical Features of GNAS Mutation Carriers with Severe Obesity. We evaluated 22 patients who presented with severe childhood-onset obesity and among whom 19 heterozygous GNAS mutations were identified, including 16 missense, 2 nonsense, and 1 frameshift mutation (Fig. S1 in the Supplementary Appendix). The R265H mutation was present in 4 unrelated patients. Some mutations were reported previously in patients with pseudohypoparathyroidism (Table S1). Nine probands inherited mutations from their mothers, 8 of whom were overweight or obese (Table S2); 8 mutations arose de novo (Table 1). All the implicated variants, some of which were originally designated as variants of uncertain significance (and were found to be pathogenic in this study),2 were submitted to the ClinVar repository under accession numbers SCV001573808 through SCV001573826.
Figure 1. Figure 1. Obesity-Associated GNAS Mutations and MC4R Signaling.
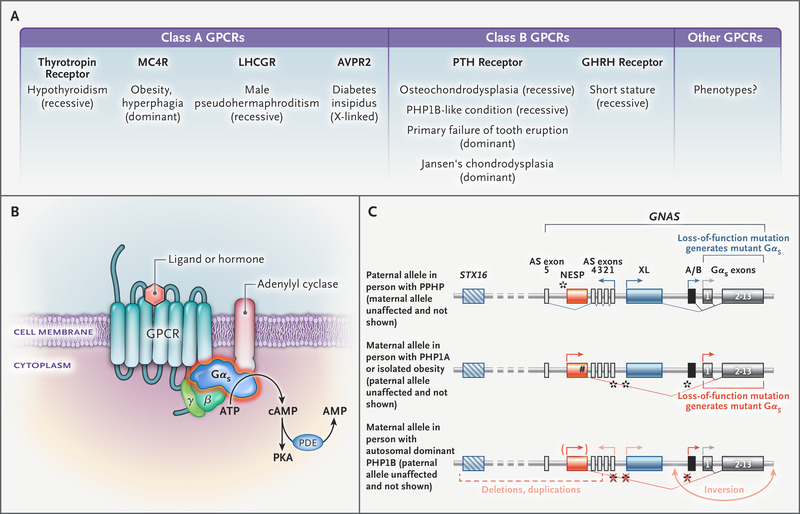
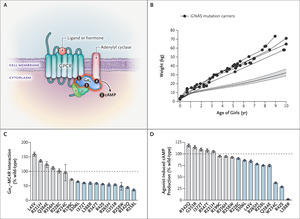
Figure 1. Obesity-Associated GNAS Mutations and MC4R Signaling. Panel A shows the following molecular mechanisms: ligand-induced coupling between stimulatory G-protein alpha subunit (Gαs) and G protein–coupled receptors (GPCRs) (1), production of cyclic AMP (cAMP) by this interaction (2), receptor-independent production of cAMP (3), and the interaction between Gαs and adenylyl cyclase (4). Panel B shows the weights of female GNAS mutation carriers. The 5th and 95th percentiles were based on data derived from the general population of the United Kingdom (dashed lines). Data on the weights of boys are included in Figure S2 in the Supplementary Appendix. Panel C shows the mean values for the interaction between melanocortin 4 receptor (MC4R) and Gαs in functional studies of wild-type and mutant forms of Gαs. Panel D shows the maximal efficacy of agonist-induced cAMP production. In Panels C and D, ? bars indicate the standard error, dashed lines wild-type Gαs, and blue bars loss-of-function mutations.
None of the patients were thought to have pseudohypoparathyroidism when referred for evaluation of severe obesity. To determine whether the implicated variants affect the function of Gαs protein, we performed experiments in Gαs-null cells. The standard assay of Gαs bioactivity cannot detect defective coupling of Gαs to GPCRs,9 so we developed a protein–protein interaction assay to study this mechanism (Figure 1A). We also measured receptor-dependent and receptor-independent cyclic AMP production and the interaction of Gαs with adenylyl cyclase 2 (Figure 1A). We studied GPCRs that mediate hormone resistance and GPCRs involved in weight regulation. The nonsense and frameshift mutations resulted in proteins that were not expressed and caused a complete loss of function. All 16 missense mutant proteins were expressed at levels similar to those of normal (wild-type) Gαs, but they had impaired coupling to GPCRs, cyclic AMP production, or both (Table S3).
GNAS Mutations and MC4R SignalingWe reviewed the clinical records of the patients and assessed features of pseudohypoparathyroidism that were present at the time of genetic diagnosis (Table 1 and Table S4). Although the mean (±SD) gestational age at birth of the GNAS mutation carriers was 39.6±1.7 weeks, the mean standard-deviation score for birth weight was low (−0.72), consistent with mild intrauterine growth restriction (Fig. S2). All the patients had accelerated weight gain in the first 6 months of life that led to severe obesity in childhood (Figure 1B).
We investigated the effect of GNAS mutations on signaling by the Gαs-coupled melanocortin 4 receptor (MC4R), which is critical to the regulation of appetite and weight and in which heterozygous loss-of-function mutations are the most common monogenic form of obesity.14 Fourteen of 16 mutations impaired the interaction between Gαs and MC4R (Figure 1C), MC4R-mediated cyclic AMP accumulation (Figure 1D), MC4R-independent cyclic AMP accumulation, or all of these. The effect of GNAS mutations on MC4R signaling is sufficient to explain early-onset obesity in the patients (Table 1), many of whom were reported to have hyperphagia, a cardinal feature of MC4R deficiency.14
Fifteen of 16 GNAS mutations impaired coupling to or signaling by β2- and β3-adrenoreceptors (Fig. S3), which mediate thermogenesis in brown adipose tissue. These findings may explain hypothermia in infancy, bradycardia, constipation, urinary retention, bronchoconstriction, and the reduced lipolytic response to epinephrine.15 Fasting insulin and glucose levels in carriers of GNAS mutations were similar to those seen in obese children in the GOOS cohort (Fig. S4).
GNAS Mutations, GHRH Receptor Signaling, and GrowthPseudohypoparathyroidism is associated with reduced growth, in part because of growth hormone deficiency caused by resistance at the level of the growth hormone–releasing hormone (GHRH) receptor.16,17 In addition, Gαs mediates the effects of PTH and PTH-related protein on chondrocytes in the growth plate.18,19
Figure 2. Figure 2. Impaired GHRH Receptor Signaling and Reduced Growth.
Figure 2. Impaired GHRH Receptor Signaling and Reduced Growth. Panels A and B show the heights of 2529 severely obese girls who were enrolled in the Genetics of Obesity Study (GOOS) and female GNAS mutation carriers, respectively; comparable data on boys are included in Figure S5 in the Supplementary Appendix. In Panels A, B, and D, dashed lines indicate the 5th and 95th percentiles according to data derived from the general population of the United Kingdom. In Panels B and D, the insets show the same data on an expanded y axis. Panel C shows mean values for the interaction between the growth hormone–releasing hormone (GHRH) receptor and Gαs in functional studies of wild-type and mutant forms of Gαs. The dashed line indicates wild-type Gαs, blue bars loss-of-function mutations, and ? bars the standard error. Panel D shows the height for female GNAS mutation carriers and the functional effects of their mutations on GHRH receptor signaling. ? bars indicate the standard error.
We plotted height data for 2270 boys and 2529 girls with severe obesity who were enrolled in GOOS. As expected, these children had a faster growth trajectory in early childhood than that indicated by the reference values derived from the general population of the United Kingdom (Figure 2A and Fig. S5). Children with GNAS mutations had accelerated growth before 12 years of age, with a mean standard-deviation score for height of 0.92; only 1 patient (with the Y163X mutation) had short stature at enrollment. In 14 of 18 children with GNAS mutations, growth trajectories before 12 years of age were similar to those in other severely obese children (Figure 2B); bone age was generally not advanced (Table S5). However, 6 of 11 GNAS mutation carriers had a reduced pubertal growth spurt, reduced final height, or both (Figure 2B and Table 1). One patient (with the S306L mutation) of 10 patients who attained final height before or during the study had short stature (Table 1).
Patients with a reduced pubertal growth spurt carried GNAS mutations that impaired GHRH receptor signaling (mean standard-deviation score for height, −0.90) (Figure 2C and 2D), whereas growth followed predicted percentiles in those with GNAS mutations that did not impair function (mean standard-deviation score for height, 0.75; P=0.02). Most GNAS mutations impaired luteinizing hormone–chorionic gonadotropin receptor signaling and follicle-stimulating hormone receptor signaling. All the patients had pubertal development, which suggested sufficient activity in the residual hypothalamo–pituitary–gonadal axis. A reduced pubertal growth spurt in some patients may indicate growth hormone deficiency, the consequences of impaired PTH or PTH-related protein signaling on the growth plate, or both. Since MC4R deficiency is associated with accelerated growth (because of hyperinsulinemia),20 impaired MC4R signaling resulting from GNAS mutations may counterbalance the negative effects of partial growth hormone deficiency on growth in early childhood.
GNAS Mutations, Thyrotropin Receptor Signaling, and Developmental Delay Figure 3. Figure 3. Impaired Thyrotropin Receptor Signaling and Elevated Thyrotropin Levels in GNAS Mutation Carriers.
Figure 3. Impaired Thyrotropin Receptor Signaling and Elevated Thyrotropin Levels in GNAS Mutation Carriers. Panel A shows mean thyrotropin levels in 522 patients in the GOOS cohort and 16 GNAS mutation carriers. Panel B shows mean thyrotropin levels in children (<10 years of age) with GNAS mutations and the functional effects of their mutations on thyrotropin receptor signaling, as compared with that in 340 severely obese children from the GOOS cohort. In Panels A and B, each circle represents an individual patient. Horizontal lines indicate means, and ? bars the standard error.
GNAS mutations can affect signaling by the Gαs-coupled thyrotropin receptor,21,22 which drives thyroid hormone synthesis. Thyrotropin resistance (a high thyrotropin level with normal or low free thyroxine [free T4] levels in the absence of goiter and antithyroid antibodies17) was evident in 11 of 16 GNAS mutation carriers (Table 1). In some instances, an elevated thyrotropin level was attributed to subclinical hypothyroidism, which is a frequent finding in childhood obesity.23 Although the mean thyrotropin level was within the normal range in 522 severely obese children in the GOOS cohort, this level was elevated in 16 GNAS mutation carriers (Figure 3A) (P<0.001). Free T4 levels were normal in children with GNAS deficiency, a finding that is consistent with compensation by the thyroid axis (Fig. S6). Carriers of loss-of-function GNAS mutations had significantly higher mean (±SD) thyrotropin levels than 340 severely obese children from the GOOS cohort (8.4±4.7 vs. 3.9±2.6 mIU per liter; P=0.004) (Figure 3B), whereas carriers of mutations with wild-type–like activity had similar thyrotropin levels.
All 17 of the GNAS mutations that were associated with developmental delay impaired thyrotropin receptor–mediated cyclic AMP production, Gαs–thyrotropin receptor interaction, or both. Some patients received levothyroxine (Table 1); however, because thyrotropin resistance was not recognized, treatment was often discontinued. Partial thyrotropin resistance may explain the low basal metabolic rate reported previously in patients with GNAS deficiency24 and in 2 of 6 patients in our study; in 1 patient with the R265H mutation, the measured basal metabolic rate was 60% of that predicted according to age, sex, and body composition, and in 1 patient with the W234C mutation, this rate was 82% of that predicted.
Poor Correlation between Skeletal Abnormalities and PTH ResistanceGNAS mutations that affect signaling by the Gαs-coupled PTH receptor lead to PTH resistance, reduced 1-alpha hydroxylation of vitamin D despite hypocalcemia, and impaired PTH-stimulated down-regulation of phosphate cotransporters, which reduce urinary phosphate excretion and cause hyperphosphatemia. PTH resistance in the proximal renal tubules can be compensated for by the actions of PTH in bone and the thick ascending renal tubule that can be mediated by G proteins other than Gαs,25 and thus it is not affected by GNAS mutations. As such, an elevated PTH level can maintain normocalcemia for some time. In our cohort, 11 patients had an elevated PTH level with normal calcium levels in early childhood (Table 1).
Figure 4. Figure 4. Subtle Skeletal Features in a Carrier of a GNAS Mutation that Impairs Parathyroid Hormone Receptor Signaling.
Figure 4. Subtle Skeletal Features in a Carrier of a GNAS Mutation that Impairs Parathyroid Hormone Receptor Signaling. Panel A shows clinical photographs of a 10-year-old patient with a GNAS R199C mutation and mild brachydactyly with subtle indentation overlying the fifth metacarpophalangeal joint. Characteristic short, broad distal phalanges of the thumbs (inset) are absent. Panel B shows the interaction between the parathyroid hormone (PTH) type 1 (PTH1) receptor and R199C Gαs. Horizontal lines indicate means. AUC denotes area under the curve. Panel C shows the dose–response curve for PTH-induced cAMP production for R199C Gαs as compared with that for wild-type Gαs and mock-transfected cells. In Panels B and C, ? bars indicate the standard error.
The majority of obesity-associated GNAS mutations impaired PTH type 1 receptor signaling. In keeping with the paucity of truncating mutations,26 only one patient (with the Y163X mutation) had subcutaneous ossifications. Ten patients had brachydactyly, which was often subtle (Figure 4A and Table 1). These findings indicate that the skeletal features of pseudohypoparathyroidism do not directly correlate with the degree of PTH resistance (Figure 4A through 4C) and may be absent in some patients with GNAS mutations that nonetheless impair signaling by the PTH receptor (Table 1 and Fig. S7).
Comments (0)