Immortalized BV2 microglia cell culture
BV2 cells were generously gifted by Derek Costello, University College Dublin. BV2 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Corning, 15–013-CM) supplemented with fetal bovine serum (FBS 10%; Gibco, 10,270,106), Penicillin–Streptomycin (1%; Gibco, 15,070,063), and HEPES (15 mM; Sigma-Aldrich, H0887-100 mL) at 37 °C with 5% CO2 saturation. The cells were routinely passaged at 80–90% confluency using trypsin. All data were derived from passages 11 through 18. BV2 microglia were seeded on surface-treated plastic (6-well plate; Greiner, 657,160) or poly-D-lysine (0.1 mg/ml; Gibco, A3890401)-coated glass. Glass coverslips (13 mm; VWR, 631–0149) were coated with 100 mL of poly-D-lysine for 24 h at room temperature.
Neural stem cell culture
Neural stem cells (NSCs) were isolated from day 14–16 mouse embryos of wildtype C57BL/6 embryos (male and female) following a previously described protocol [27] and cultured in Dulbecco’s Modified Eagle Medium (DMEM) and Ham’s F-12 Nutrient Mixture (DMEM/F12; Corning, 10–092-CV) supplemented with B27 (1X; Gibco, 17,504,044), Penicillin–Streptomycin (1%; Gibco, 15,070,063), and fungizone (0.2%; Gibco, 15,290,018). NSCs were passaged at a ratio of 1:3 every 7 days using accutase (Gibco, A1110501) for cell detachment. NSCs were seeded in fibronectin (10 mg/ml; Bio-Techne, 1030-FN-05 M) coated 96 well PhenoPlates (Revvity, 6,055,302) at a density of 31,250 cells/cm2 for 2 days prior to co-culture with miPSCs.
Murine iPSC-derived microglia culture
Mouse embryonic fibroblasts (MEFs) and CX3CR1eGFP/ + CCR2RFP/ + miPSCs were gifted by Prof. Peter Ponsaerts, University of Antwerp, Belgium. Irradiated MEFs were cultured in DMEM (Corning, 10–013-CV) supplemented with FBS (10%; Gibco, 10,270,106), Penicillin–Streptomycin (1%; Gibco, 15,070,063), and Minimum essential medium, Non-Essential Amino Acids Solution (1X MEM-NEAA; Gibco, 11,140,035) at 37 °C with 5% CO2 saturation. Irradiated MEFs were seeded in surface treated 6-well plates (Greiner, 657,160). After 24 h, miPSCs were seeded on top of the irradiated MEFs and cultured in Knock-out DMEM (KO-DMEM; Gibco 10,829,018) supplemented with Embryonic stem cell FBS (ESC-FBS 15%; Gibco 10,439,001), Penicillin–Streptomycin (1%; Gibco, 15,070,063), L-Glutamine (2 mM; Gibco, 25,030,024), MEM-NEAA (1X; Gibco, 11,140,035), 2-mercaptoethanol (0.24 mM; Gibco, 31,350,010), and Recombinant mouse Leukemia Inhibitory Factor (LIF 106 U/ml; Sigma-Aldrich, ESG1106). After two days in culture, miPSCs were passaged 1:5 following detachment using 0.05% Trypsin–EDTA (Gibco, 25,300,054) and seeded onto a new layer of irradiated MEFs. Murine iPSCs were generated following the protocol previously described [27]. Briefly, 500,000 miPSCs were seeded per agarose-coated 10 cm petri dishes in microglia differentiation medium (MDM) consisting of Glasgow’s minimum essential medium (GMEM; Gibco, 21,710,025) supplemented with FBS (10%; Gibco, 10,270,106), Penicillin–Streptomycin (1%; Gibco, 15,070,063), MEM-NEAA (1X; Gibco, 11,140,035), Sodium pyruvate (1 mM; Gibco, 11,360,039), and 2-mercaptoethanol (0.1 mM; Gibco, 31,350,010). Embryoid bodies (EBs) formed were transferred to new agarose-coated dishes on day 4. On day 8, the EBs were transferred into gelatin-coated (0.1% in sterile dH20 for 1 h; Sigma-Aldrich, G1393-20ML) T175 flasks in MDM supplemented with SCF (200 ng/ml; ImmunoTools, 12,343,327) and VEGF-A (10 ng/ml; ImmunoTools, 12,343,663). On day 11, the medium was changed to MDM supplemented with 15% (v/v) L-929 conditioned media, 20% (v/v) bEND5 conditioned media, IL-3 (2 ng/ml; ImmunoTools, 12,340,033), and GM-CSF (40 ng/ml; PeproTech, 315–03). On day 15, the media was changed back to the original MDM. From days 29–57, GFP⁺ floating cells called primordial microglia progenitors (PMPs) released by the EBs were harvested and cultured at a density of 31,250 cells/cm2 on a layer of NSCs for 7 days in MDM as described previously by Quarta [27] and Haenseler et al., [28] who demonstrated that this window supports microglial marker expression in a neuroectodermal setting. These NSC/miPSC microglia cultures were used for stimulation experiments, subsequent enzyme-linked immunosorbent assay (ELISA) and/or immunocytochemistry experiments.
Cell treatments
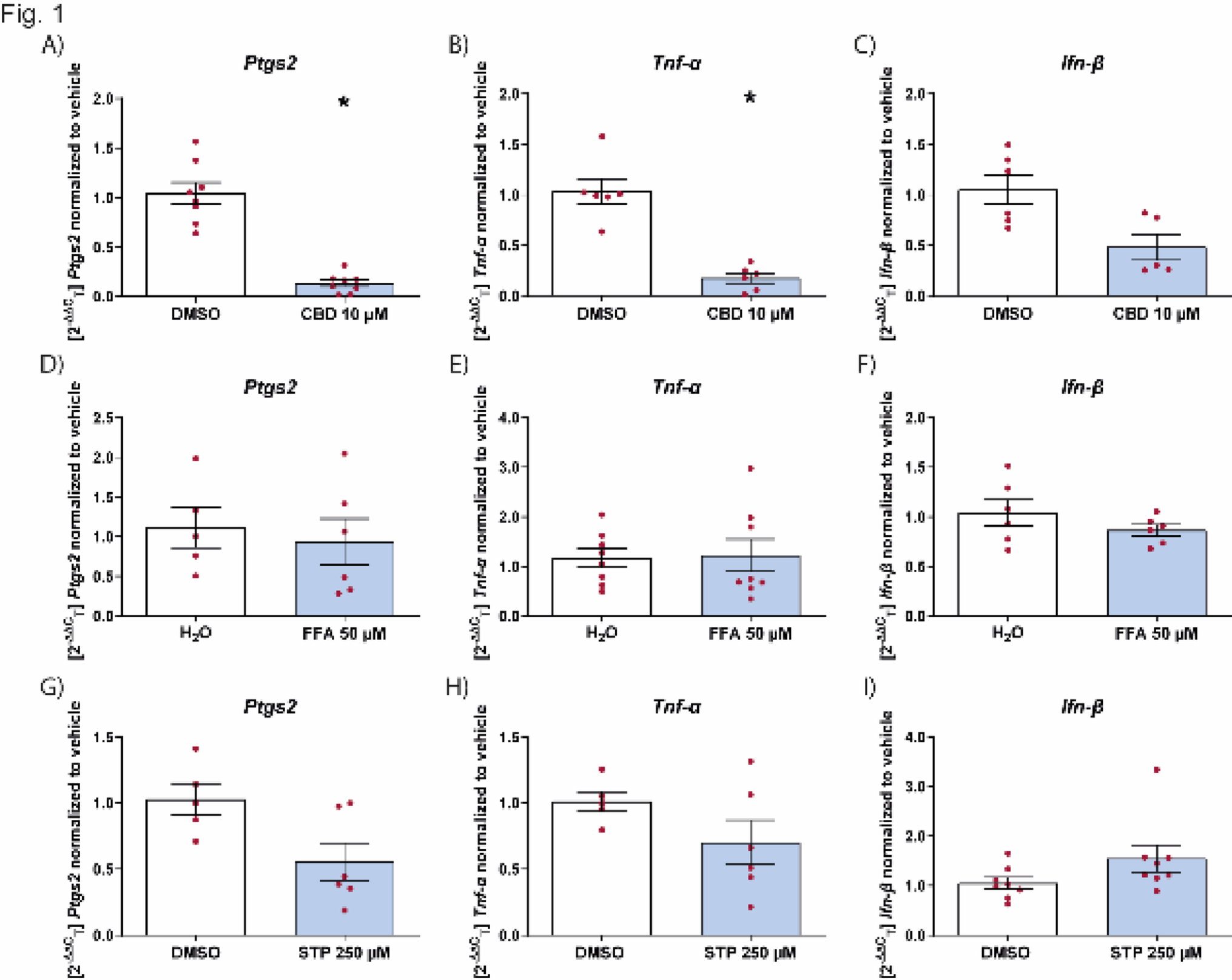
To investigate inflammatory responses in microglia, BV2 cells were treated for 24 h with LPS (InvivoGen, tlrl-b5lps) and/or IL-13 (ImmunoTools, 12,340,137) at concentrations of 1, 10, 100 or 1000 ng/ml and 20, 100, or 500 ng/ml, respectively. Cells were starved in 0.5% FBS during treatment. For timed dual stimulation experiments, BV2 cells were pre-treated with LPS (100 ng/ml) for 24 h before being subsequently treated with either low serum media control, LPS (100 ng/ml), or IL-13 (20 or 100 ng/ml) for an additional period of 6, 24, or 48 h. To investigate inflammatory responses in miPSC-microglia, the NSC/miPSC microglia co-cultures were treated for 24 h with LPS (InvivoGen, tlrl-b5lps) and/or IL-13 (ImmunoTools, 12,340,137) at concentrations of 1, 10, or 100 ng/ml and 20, 100, or 500 ng/ml, respectively; treatments were prepared in MDM.
To determine how matrix proteins impact IL-13 response, BV2 cells were grown in 6-well plates coated with collagen IV (10 mg/ml; Bio-Techne, 3410–010-02) or fibronectin (10 mg/ml; Bio-Techne, 1030-FN-05 M). Plates were coated for 2 h and then washed three times with sterile 1X DPBS without Ca2+ and Mg2+ (Gibco, 14,190,144). To assess how hyperkalemia or excessive glutamate impacts IL-13 response in microglia, BV2 or miPSC-microglia cells were treated in media supplemented with either [K+]e (Fisher Chemical, P/4240/60), 15 mM or 35 mM final concentration for BV2 cells and 10 mM or 20 mM final concentration for miPSC-microglia or glutamate (Sigma-Aldrich, G1626-100G), 2 mM or 20 mM final concentration. To assess the polarising ability of IL-13 on BV2 microglia under acidic conditions, BV2 growth media was adjusted from a pH of 7.3 to a pH of 6.7 and sterile filtered before use with 0.22 μm syringe filter (Merck, SLGP033RS).
When assessing the impact of matrix proteins, potassium, glutamate and pH on IL-13 response, BV2 cells were seeded at a density of 200,000 cells per well and left to proliferate for 24 h in growth media. Following this, the cells were starved with 2% serum-containing media for another 24 h before being treated with IL-13 (100 ng/ml, for 24 h), prepared using 2% FBS-containing media. The cells were then lysed with TRIzol for RT-qPCR analysis. PMPs were cultured at a density of 31,250 cells/cm2 on a layer of NSCs for 7 days in MDM. Following this, NSC/miPSC- microglia were treated for 24 h with IL-13 (100 ng/ml).
Enzyme-linked immunosorbent assay
The concentration of TNFα in the cell supernatants were determined using ELISA according to the instructions provided by the manufacturer (Bio-Techne, DY410-05).The culture supernatant was collected from the wells of a 6 or 24 well plate that had been seeded with BV2 cells and treated with LPS and/or IL-13, or 96 well PhenoPlates seeded with NSC/miPSC-microglia treated with LPS and/or IL-13, [K+]e (10 mM or 20 mM final concentration) or glutamate (2 mM or 20 mM final concentration). A 96 well plate was coated with the capture antibody diluted in plate coating buffer (800 ng/ml), sealed, and incubated overnight. Following the overnight incubation, the wells of the plate were aspirated and washed with wash buffer thrice. The wells were subsequently blocked with BSA (1% in PBS) for 1 h at room temperature. The aspiration and wash step were repeated. 100 ml of the supernatants was added to each well of the 96-well plate and incubated for 2 h at room temperature. The aspiration and wash step were repeated. The plate was subsequently incubated with detection antibody diluted in reagent diluent (37.5 ng/ml) for 2 h at room temperature. The aspiration and wash step were repeated. Following this, the plate was incubated with streptavidin–horseradish peroxidase (40-fold dilution in PBS with 1% BSA) for 20 min in the dark at room temperature. The final aspiration and wash step were repeated. Substrate reagent was added to the wells and incubated for 20 min at room temperature. Following this, sulfuric acid (2N) was added to stop the reaction. Immediately after, the optical density was measured at 450 nm and 570 nm a microplate reader (Molecular Devices, 89,212–396 (PLUS 384)). The readings at 570 nm were subtracted from the readings obtained at 450 nm prior to analysis and analyte concentration was determined in relation to known TNFα standards.
RNA isolation and quantitative real-time PCR
Total RNA was extracted from BV2 microglia using TRIzol (Thermo Fisher Scientific, 15,596,018). Cells were gently washed using ice-cold 1X DPBS prior to lysis with TRIzol. Following lysis, RNA was isolated via phase separation with chloroform, precipitation with isopropanol, and after two washes with 75% ethanol, the RNA pellet was dissolved in RNase-free H2O. 2 μl of GlycoBlue (ThermoFisher, AM9516) was added as a co-precipitant. The concentration of RNA was measured using a Spectrophotometer (DeNovix, DeNovix DS-11). Sufficient sample purity was accepted at values of > 1.8 for the 260/280 ratio and > 2.0 for the 260/230 ratio. cDNA was synthesised from total RNA using SuperScript™ II Reverse Transcriptase (Thermo Fisher Scientific, 18,064,014). All quantitative reverse transcription polymerase chain reactions (qRT-PCR) were performed in 384-well plates on a QuantStudio™ 7 Flex Real-Time PCR System (Thermo Fisher Scientific, 4,485,701) using SYBR Green PCR Master Mix Reagent (Thermo Fisher Scientific, 4,364,344) with Quantstudio Real-Time PCR software. Peptidylprolyl Isomerase A (PPIA) was used to normalise expression levels. The 2−ΔΔCT method was used to calculate relative changes in gene expression. The sequences used for the primers are shown in Table 1.
Table 1 Primers for RT-qPCRScratch wound assay
To assess infiltration of wound areas by microglia, BV2 cells were grown for 2 days in DMEM supplemented with (10%; Gibco, 10,270,106), Penicillin–Streptomycin (1%; Gibco, 15,070,063), and HEPES (15 mM; Sigma-Aldrich, H0887-100 mL), cultured at 37 °C with 5% CO2 saturation as above, and treated with either IL-13 or LPS. After 24 h, when the monolayer is typically ~ 90% confluent, a ~ 250 µm scratch was made. The cells were gently washed with DMEM and low-serum media containing treatments (LPS or IL-13) was then added. Infiltration was determined 24 h later, using image binarisation to determine the relative change in scar density as compared to untreated controls.
Immunocytochemistry
To perform immunocytochemistry (ICC), cells were first washed in PBS on a shaker for 10 min at room temperature. The cells were then blocked for 30 min at room temperature using protein block (Abcam, ab64226) After blocking, the primary antibodies, Anti-MHCII (Abcam, ab180779), Anti-Iba1 (FujiFilm Wako, 019–19741), Anti-Arginase 1 (Abcam, ab91279) and Anti-F4/80 (BioRad, MCA497GA) were added at a dilution of 1:400 using an antibody diluent consisting of 1% protein block and 0.05% Triton-X in 1X PBS, and incubated with the cells overnight at 4 °C. Next, the cells were washed three times for 5 min each in 1X DPBS on a shaker in the dark. The secondary antibodies Goat anti-rabbit 568 (Invitrogen, A11011) and, Goat anti-rat 647 (Invitrogen, A21247) were added at a dilution of 1:400 and incubated with the cells for 2 h at room temperature in the dark. The cells are again washed three times for 5 min each in 1X PBS on a shaker in the dark. When performing high-content screening, BV2 cells were stained with a green cell mask (Invitrogen, C37608) for 30 min at room temperature, to label the cell membrane for morphological analysis, the cells were then washed with 1X PBS. To visualise the nuclei, the cells were counterstained with 300 nM Hoechst 33342 for 10 min at room temperature in the dark. The cells were washed thrice with 1X PBS and subsequently kept in PBS prior to imaging on an Olympus FV1000 microscope (using 405 nm, 488 nm and/or 559 nm laser-scanning confocal imaging) or an Opera Phenix High-Content Screening optical system (see below).
Calcium imaging
To register live calcium activity in microglia, BV2 cells were grown in complete DMEM on poly-D-lysine-coated 35 mm glass confocal dishes for two days as above. Media was exchanged for low-serum (2%) complete DMEM and cells were treated with either LPS or IL-13. The cells were washed three times with warmed sterile PBS and then incubated with Fluo4-AM (Thermo Fisher Scientific, F14201) in Hanks’ balanced salt solution (HBSS) (Sigma-Aldrich, D8537) for 1 h, 37 °C. Cells were washed once and imaged at 37 °C in artificial cerebrospinal fluid (aCSF, in mM; NaCl, 125; KCl, 2.5; NaH2PO4, 1.25; NaHCO3, 11.9; CaCl2, MgSO4∙7H2O, 1.3; D-glucose, 18; HEPES, 10). Calcium imaging was performed using a Nikon eclipse TS100 brightfield microscope with a custom-fitted fluorescence microscopy module. A 470 nm LED (Thorlabs, M470L5) was mounted to the rear optical port and routed through a FITC-filter cube, consisting of a 475/35 nm bandpass (excitation) filter (Thorlabs, MF475-35), a 530/43 nm bandpass (emission) filter (Thorlabs, MF540-43), and a 498 nm longpass dichroic mirror (Thorlabs, MD498). Fluorescent images were acquired at 2 Hz using an Optika CP-6 digital camera (Optika Italy, 57,549) and a Nikon Plan Fluor 20X 0.45 NA objective. Calcium imaging performed here is as described in [29].
High-content screening
To examine the expression of anti-inflammatory markers by BV2 or NSC/miPSC-microglia cells in response to pro- and anti-inflammatory stimuli, immunofluorescence and high content screening were carried out using the Opera Phenix high-throughput confocal microscope (Revvity, HH14001000). Fluorescence intensity and cell morphology were analysed using the Harmony software (Harmony® high-content analysis software). For BV2 cell analysis, 96-well optical plate (Revvity, 6055302) was coated with 100 ml Poly-D-Lysine (0.1 mg/ml, Gibco, A3890401) for 4 h at room temperature. The plate was subsequently washed with PBS thrice. 10,000 BV2 cells were seeded per well and left to proliferate for 48 h. Following this, the cells were starved with 2% serum media for another 24 h. Subsequently, BV2 cells were treated with IL-13 (100 ng/ml), LPS (100 ng/ml), TGFβ1 (50 ng/ml; Biolegend, 763,104), TGFβ2 (50 ng/ml; Biolegend, 583,301), M-CSF (10 ng/ml; Peprotech, 315–02), and/or ovine cholesterol (Avanti Polar Lipids / Merck Millipore, 700000P). After 24 h of treatment, the cells were fixed with 2% paraformaldehyde (PFA) for 20 min. The cells were washed thrice with DPBS for 5 min before and after fixation. PMPs were developed on a layer of NSCs for 6 days, on day 7, the NSC/miPSC-microglia were treated with IL-13 (100 ng/ml), LPS (100 ng/ml), TGFβ1 (50 ng/ml), TGFβ2 (50 ng/ml), M-CSF (10 ng/ml), and/or ovine cholesterol. Alternatively, NSC/miPSC-microglia were treated with KCl (10 mM or 20 mM final concentration [K+]e) or glutamate (2 mM or 20 mM final concentration) to quantify the effects of microenvironmental factors on pro- or anti-inflammatory marker expression. Following 24 h of treatment, the cells were washed once with DPBS, fixed for 20 min using 2% PFA, and then washed thrice with DPBS.
Statistical analysis
A minimum number of three experimental replicates with at least two technical replicates was used in all experiments unless otherwise noted. For the statistical tests the following analyses of variance were used; Unpaired t-test, one-way ANOVA with Šídák’s or Dunnett’s multiple comparison tests; two-way ANOVA with Benjamini, Krieger and Yekutieli False Discovery Rate post-hoc test. Results were considered significant at P < 0.05. Statistical significance was denoted by *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.







Comments (0)