
Remember me

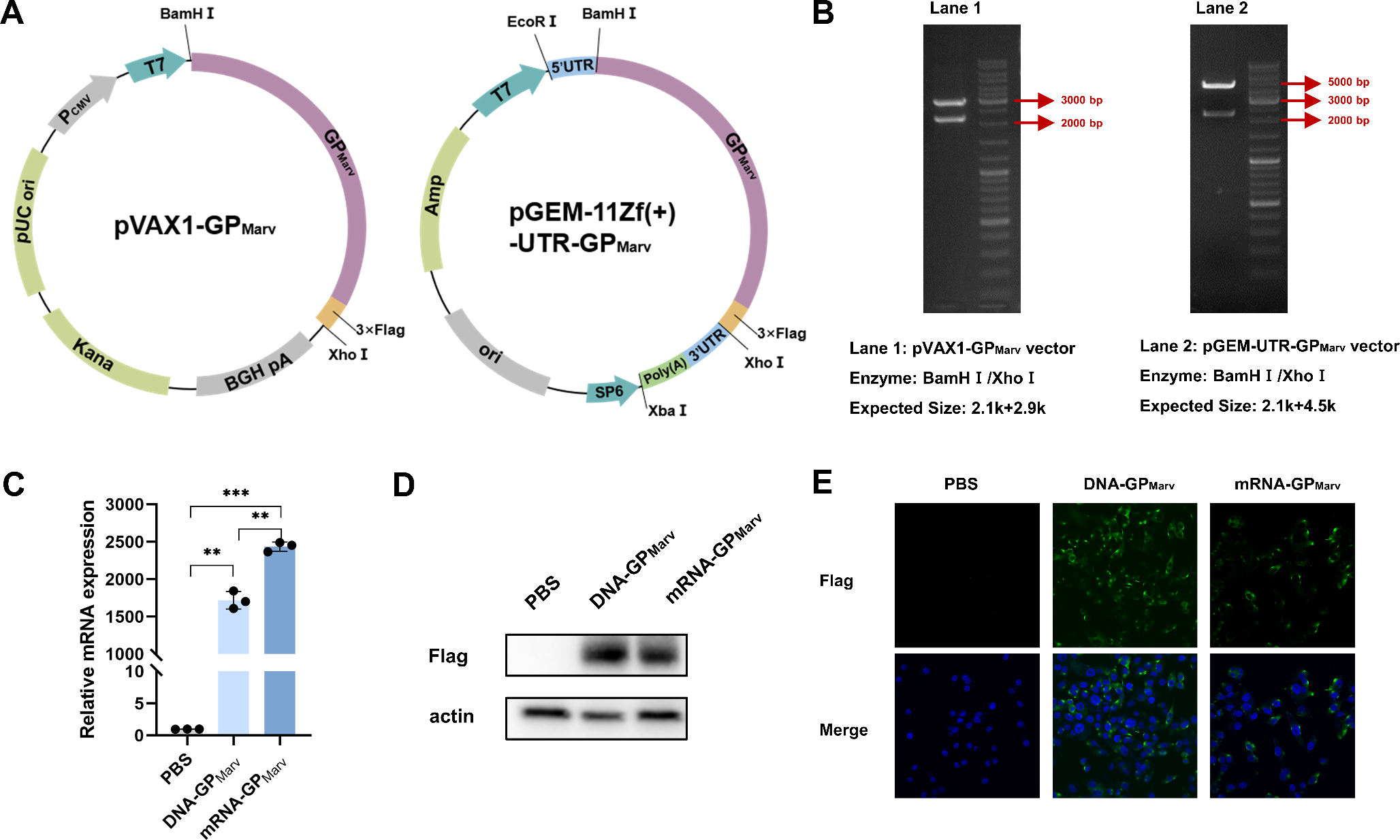

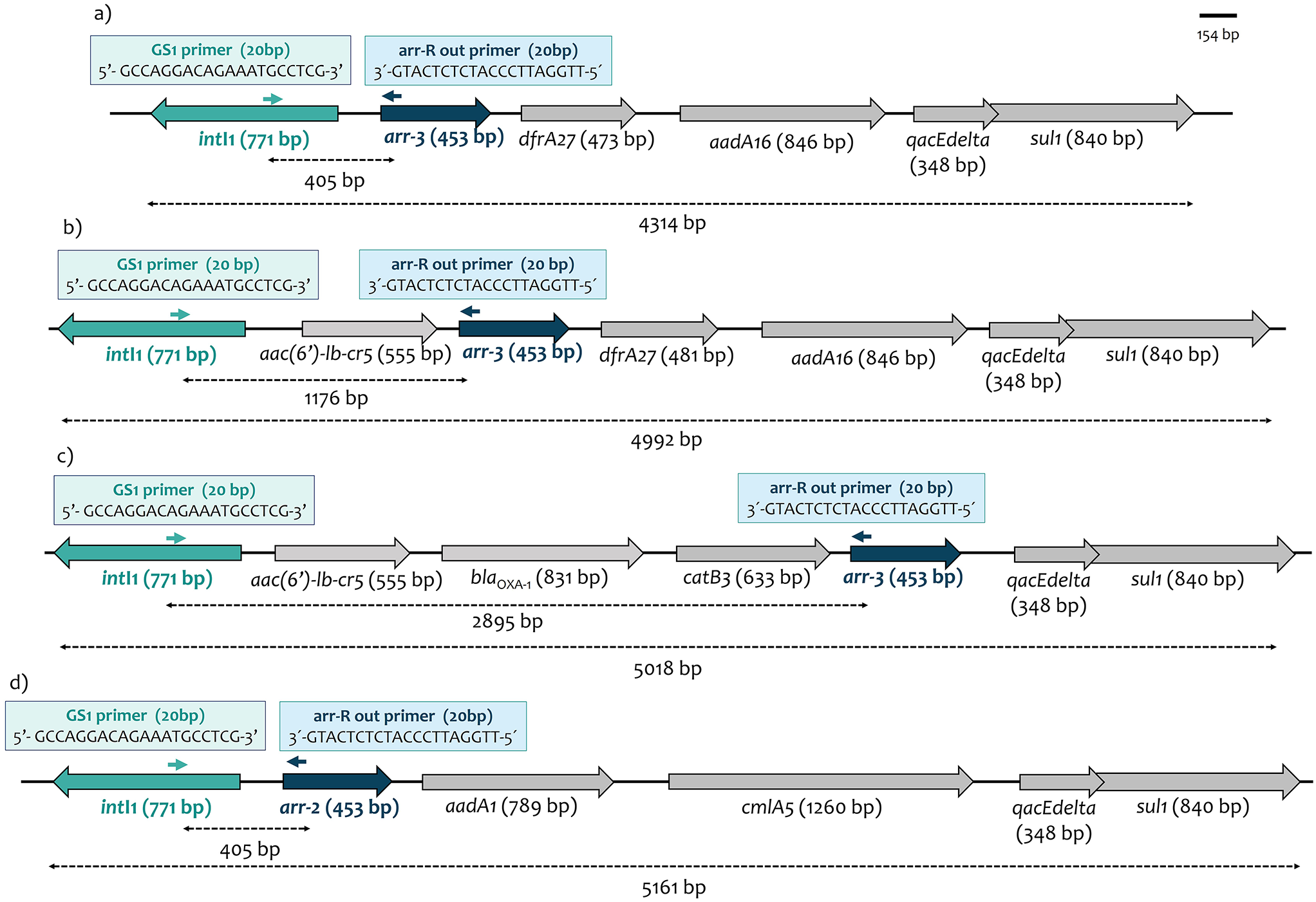

Our study included patients with pneumococcal CAP during the acute infection phase, with nasopharyngeal samples collected at the time of diagnosis and admission to the hospital. The same patients were sampled again three months post-infection. Matched asymptomatic control subjects were also analyzed for comparison. In total, 61 patients with CAP and 61 healthy controls were included (Table 1). COPD, CAD, and previous or current smoking were significantly more common among CAP patients compared to controls.
Table 1 Cohort descriptionFor clinical diagnosis, specific PCR tests from nasopharyngeal samples for S. pneumoniae (targeting the lytA gene) and other potential pathogens, along with standard microbiological cultures (nasopharynx, lower respiratory tract and blood) and urine antigen tests, were performed (Table 2). Patients who tested positive for S. pneumoniae in any test were included in this study (Table S2). Samples from these patients were additionally analyzed using 16 S rRNA (V4 region) amplicon sequencing for microbiome investigation. This analysis revealed Streptococcus spp. in all but one nasopharyngeal sample, with feature counts (FC) ranging from 54 to 154,588 (median 2,697). The specific PCR test for S. pneumoniae (lytA) was positive in 58% of the samples tested. Notably, all samples containing more than 10,000 FC of Streptococcus spp. were positive in lytA PCR. Additionally, pneumococci were detected in 16% of the nasopharyngeal cultures. Most patients (69%) tested positive for the pathogen using the UAD, while 44% were positive with the UAT. Blood cultures identified the bacterial species in 16% of the cases. However, despite all patients being diagnosed with S. pneumoniae infection, none of the tests were positive for all samples. Serotyping of available isolates (n = 45) revealed that the most common serotypes of included patients were serotype 3 (27%) followed by 8 (11%) and 9 N (11%) (Table S3).
Table 2 Microbiological diagnostic testingPost-infection nasopharyngeal Microbiome showed reduced bacterial biomass, increased alpha diversity, and lower Microbiome stability compared to infection and control groupsNasopharyngeal samples from patients with S. pneumoniae CAP were compared with a matched control group of healthy volunteers. Additionally, patient samples collected at different time points were also included in the analyses. Absolute quantification of the 16 S rRNA gene copies showed no statistical differences between the nasopharyngeal bacterial biomass of pneumococcal pneumonia patients and matched control samples (Fig. 1). In contrast, significant lower bacterial load was found in post-infection samples compared to those collected during infection (p = 0.004).
Fig. 1
Bacterial biomass of nasopharyngeal swab samples. Scatter dot plot depict the quantitative analysis of bacterial biomass measured by qPCR, represented as 16 S rRNA gene copies per sample. Statistical significance was assessed using pairwise comparisons with the Wilcoxon test, with p-values < 0.01 indicating significant differences
To evaluate microbiome diversity within the samples, different metrics examining taxa richness and evenness were calculated. These included, Observed OTUs: measures the total number of taxa present in a sample, Shannon index: accounts for both richness and evenness, with a stronger emphasis on richness, and the Simpson index: similar to Shannon but places greater weight on evenness. The comparison between samples showed that nasopharyngeal samples from the infection phase and the control group showed no significant differences in the amount of taxa observed (richness) and abundances (evenness) measured (Observed OTUs p = 0.9, Shannon p = 0.91, Simpson p = 0.92) (Fig. 2A). In contrast, α-diversity indices differed significantly between infection-phase and post-infection samples (Observed OTUs: p = 0.0035, Shannon: p = 0.012, Simpson: p = 0.03). This indicates that post-infection samples had a higher number of species and a more even distribution of abundances compared to infected patients. Notably, the same results were observed when comparing post-infection samples with those from control individuals (Observed OTUs p = 0.0019, Shannon p = 0.011, Simpson p = 0.022). To assess for microbiome stability, we performed co-occurrence network analyses of significant bacterial correlations of each sample group and calculate the modularity index. This measurement indicates how well a network can be divided into distinct subgroups or modules, interpreting higher values (M = 0.4) as stable ecosystems [20]. Post-infection samples showed lower modularity (M = 0.296), as compared to patients infected samples (M = 0.341) suggesting lower microbial resilience. In contrast, the healthy microbiome exhibited the greatest stability (M = 0.429) (Figure S4).
Analysis of beta diversity, estimating for differences in microbiome diversities between groups, revealed significant differences between patient samples during infection and healthy controls (p = 0.002) (Fig. 2B). In contrast, post-infection samples showed similar beta diversity distances between these two groups (Fig. 2C). Moreover, Bray-Curtis distances indicated significant differences in microbiome diversity between samples collected during infection and post-infection phases (p = 0.004) (Fig. 2D).
Fig. 2
Diversity measurements between nasopharyngeal samples from S. pneumoniae CAP patients and healthy controls. (A) Box plots showing different indices for alpha diversity measurements of nasopharyngeal swab samples from Streptococcus pneumoniae CAP patients and healthy subjects. Comparisons are made between different sample collection time points and healthy controls. P-values from pairwise comparisons are derived using the Mann-Whitney U-test. (B-C-D) Bray Curtis distances were calculated to assess for the comparison of microbiome diversities between groups (beta diversity). Sample distances were plotted as non-metric multidimensional scaling (NMDS) plot. P-values from pairwise comparisons are determined using PERMANOVA and the permutation test for beta dispersion (betadisper). * indicates p < 0.05
Taxonomic analysis revealed similar bacterial community structures between pneumonia patients and healthy controls, with notable interindividual variability and a significant decrease in Streptococcus spp. Abundance post-infectionAnalysis of the taxonomic bacterial composition of nasopharyngeal swab samples from individuals and negative controls revealed similar taxonomic profiles and microbiome diversities (Figure S5 and S6). To improve accuracy of our microbiome analysis, we developed a custom bioinformatics pipeline that detects and eliminates contaminants found in negative controls from the dataset.
Bacterial taxonomic analysis of the filtered dataset revealed a high similarity in bacterial communities among samples from S. pneumoniae pneumonia patients during infection, post-infection, and healthy controls (Fig. 3A). The genera Corynebacterium spp., Streptococcus spp., Staphylococcus spp., Moraxella spp., and Dolosigranulum spp. were the dominant bacterial taxa in the nasopharyngeal microbiome across all analyzed samples (Table S4). Figure 3B illustrates the median abundance of these five genera across the groups. We observed high interindividual variation within groups, with samples exhibiting both high and low feature counts (FC) for the same bacterium. Notably, only the FC of Streptococcus spp. decreased progressively from the infection phase (median FC: 2,697) to post-infection (median FC: 375), compared to healthy individuals (median FC: 706).
Due to the high interindividual variation observed in bacterial communities across different groups (Figure S7A), we explored taxonomic differences among the entire sample set. An analysis of compositions of microbiomes with bias correction (ANCOMBC) was performed. This statistical test revealed a significantly increased abundance of Streptococcus spp. in patient samples during infection compared to post-infection and healthy nasopharyngeal samples, among other findings (Table S5). Since amplicon sequencing of the V4 region cannot precisely identify bacterial species, we used clinical diagnostic data to correlate detected Streptococcus spp. with diagnosed Streptococcus pneumoniae. Pearson correlation analysis found a significant association between the relative abundance of Streptococcus spp. and S. pneumoniae PCR test results in infected patient samples (Table S6), indicating that Streptococcus spp. detected through 16 S rRNA amplicon sequencing in samples from patients during infection is likely to be Streptococcus pneumoniae.
Fig. 3
Taxonomic composition of nasopharyngeal bacterial communities in S. pneumoniae CAP patients and healthy controls. (A) Mean relative abundances (RA) of the 20 most abundant genera are illustrated for each group. (B) Violin plot showing the median RA of the five most representative bacterial genera across the three study groups, with ANCOMBC statistical significance indicated. (C) Mean relative abundance of the top 10 bacterial taxa in each pneumococcal CAP sample group, classified according to the abundance of Streptococcus spp. (Strep.): Group 1 (Strep. > 50% RA), Group 2 (Strep. 50%-5% RA), and Group 3 (Strep. <5% RA)
Distinct significant bacterial associations in the nasopharyngeal Microbiome of Pneumococcal CAP patients during infection and post-infection compared to healthy subjectsGiven the high interindividual variance within this cohort but the significantly increased RA of Streptococcus spp. during infection, nasopharyngeal samples from pneumonia patients suffering from S. pneumoniae at this acute stage were categorized based on the RA of Streptococcus spp., likely Streptococcus pneumoniae. In Figure S7B, Group 1 included patients with > 50% RA. Group 2 contained samples with 50%-5% RA, and, finally, Group 3 patients with < 5% RA of Streptococcus spp. Figure 3C displays the mean RA of the 10 most abundant bacterial taxa in each group. Group 1 showed Streptococcus spp. as the dominant taxon (mean RA 79.5%) while other taxa, like Corynebacterium spp. showed to be less (mean RA 2.1%). In contrast, Corynebacterium spp. emerged as the predominant genus in Groups 2 and 3, with mean RAs of 28.2% and 30.9% respectively, while Streptococcus spp. showed decreased mean RA of 14.4% and 1.8%.
To better understand the microbial interplay between Streptococcus spp. and other bacterial taxa during infection, post-infection, and healthy states, we selected for bacteria with a mean relative abundance (RA) of more than 1% and performed Pearson Correlation Coefficient (PCC) analysis (Table S7). To enhance data visualization, we also conducted a network analysis of significant bacterial associations (Fig. 4). During the infection phase, Corynebacterium spp. showed significant negative correlations with both Streptococcus and Staphylococcus spp. Interestingly, this antagonistic relationship between Corynebacterium spp. and Staphylococcus spp. was also observed in healthy samples. Additionally, in healthy individuals, significant negative correlations were found between Corynebacterium spp. and Moraxella spp., as well as between Dolosigranulum spp. and Staphylococcus spp. In the post-infection phase, significant positive correlations were observed between Streptococcus spp. and the bacterial taxa Prevotella spp. and Veillonella spp. indicating a shift in microbial dynamics post-infection. Conversely, Corynebacterium spp. was negatively associated with Prevotella spp.
Staphylococcus spp. exhibited several significant associations with other bacteria across the different phases. During the infection phase, it showed a significant positive correlation with Lawsonella, whereas in healthy controls, it had significant negative correlations with Moraxella and Dolosigranulum. On the other hand, Bacillaceae consistently showed significant positive correlations with other bacteria depending on the phase: during infection, it correlated positively with Anaerococcus and Anoxybacillus, whereas in healthy controls, it showed positive correlations with Veillonella and Streptococcus spp.
In the post-infection phase, Corynebacterium spp. developed significant negative correlations with Prevotella spp. and Bacillaceae. Anaerococcus continued to show significant positive correlations with Prevotella and Peptoniphilus. Notably, Streptococcus spp., which had significant negative correlations with Corynebacterium spp. during the infection phase, shifted to significant positive correlations with Veillonella and Prevotella spp. in the post-infection phase.
Fig. 4
Network visualization of Pearson Correlation Coefficients (PCC) among bacterial taxa in nasopharyngeal samples. The network diagram illustrates Pearson correlation coefficients (PCC > 0.1) for bacterial taxa with a mean relative abundance greater than 1% in nasopharyngeal swab samples from both S. pneumoniae CAP patients and healthy controls. Positive PCCs are depicted in red, while negative PCCs are shown in blue. Significant positive and negative associations are represented by thick lines, while non-significant associations are shown with thin lines. A positive bacterial association suggests that both bacteria are likely to be present together in a sample, while a negative correlation indicates that if one bacterium is present, the other might be absent
These findings reveal intriguing shifts in microbial interactions across infection, post-infection, and healthy states. To explore these bacterial interactions further, we conducted a focused literature search. Using keywords such as “bacterial association” and/or “bacterial correlation,” we reviewed relevant studies on bacterial interactions within the nasopharyngeal microbiome of adults with pneumococcal pneumonia up to August 2024. Our search included literature on interactions in the same or other respiratory sites, as well as other body areas, and examined both infection and healthy states, with a particular focus on adults and children (Table S8). Interestingly, many of the significant bacterial associations identified in the nasopharynx in this study have not been previously described. Similar associations have been reported in other parts of the body, such as the lungs, oral cavity, and skin, but not specifically in the nasopharynx. For example, positive associations between Streptococcus spp. and Veillonella spp. have been observed in the oral cavity, while positive correlations between Streptococcus and Prevotella spp. have been noted in the lungs. Another notable finding is the negative association between Corynebacterium and Streptococcus spp., which has been documented in the nasopharynx of healthy children but not in adults. Similarly, the negative association between Corynebacterium and Moraxella spp. has been observed in infants but not in adults.
Correlation analysis revealed a significant association between Streptococcus spp. Abundance and viral co-infection in the nasopharynx of Pneumococcal pneumonia patientsTo further investigate the presence of Streptococcus spp. in the nasopharyngeal microbiome of these patients, we performed Pearson correlation analysis using key clinical data. This analysis allowed us to examine associations between S. pneumoniae and clinical comorbidities, such COPD and CAD, as well as smoking, which were more prevalent in CAP patients than in healthy controls (Table 1). By integrating these variables, we aimed to assess their relationship with microbiome composition and determine how these comorbidities correlate with the presence of S. pneumoniae, potentially influencing disease progression.
Given that viral infections, such as influenza A can be exacerbated with a pneumococcal superinfection with an increased risk of severe disease progression, we included the results of viral tests in our model. Additionally, other relevant clinical variables, such as length of hospital stay (LOS), contact with children, disease severity (measured by the pneumonia severity index [PSI] score), and the presence of invasive disease (indicated by S. pneumoniae -positive blood culture results), were also considered in this analysis (Table S9).
Of all variables analyzed, only viral co-infection was found to be significantly associated (p = 0.001) with the presence of Streptococcus spp. in the nasopharynx. Notably, we did not find any correlation between the relative abundance of Streptococcus spp. in the nasopharynx and the occurrence of invasive disease. Further analysis of bacterial taxonomic communities revealed a higher relative abundance of Streptococcus spp. (mean RA 32.8%) in patients with viral co-infections compared to those without (mean RA 8.7%) (Fig. 5). In contrast, Corynebacterium spp. was the dominant taxon in patients without viral co-infection (mean RA 30.6%), whereas its mean relative abundance was lower in those with positive viral tests (mean RA 15.2%).
Fig. 5
Bacterial taxonomic profiling of patients´ nasopharyngeal swabs collected during infection and grouped based on the presence or absence of a viral co-infection
Comments (0)