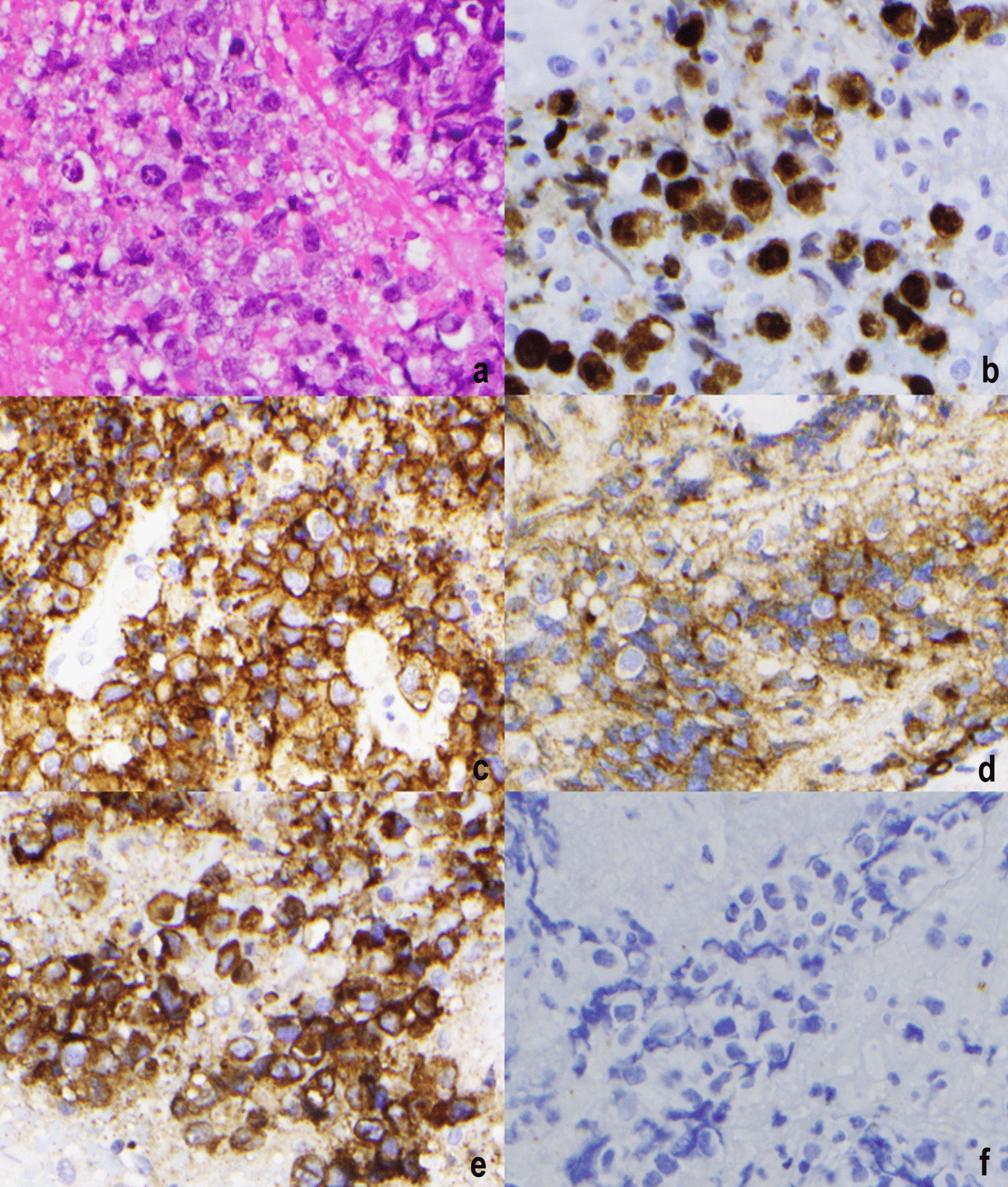
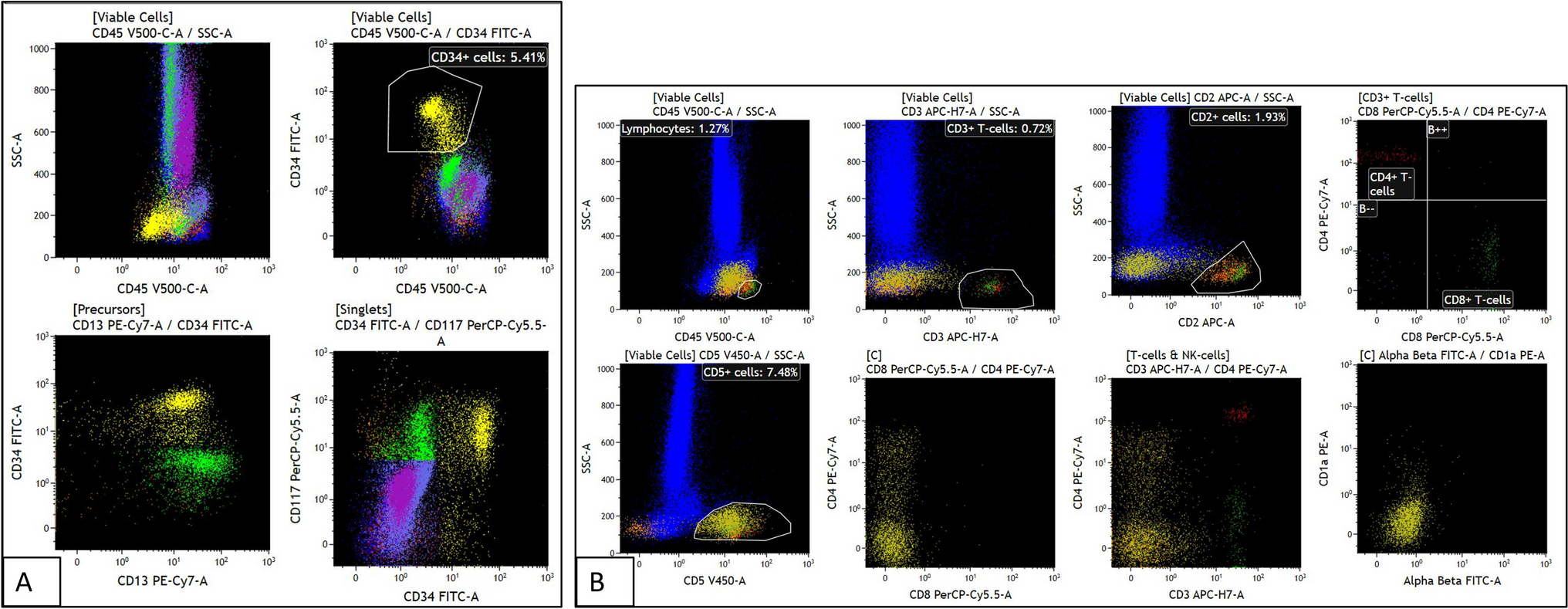
MM is a highly heterogeneous disease characterized by diverse genetic abnormalities, clinical presentations, and treatment responses [11, 12]. The disease is driven by primary events, such as hyperdiploidy and IGH locus translocations, as well as secondary events, including 1q21 gains/amplifications, 17p deletions, and MYC gene translocations [10]. The most frequent IGH translocations—t(11;14), t(4;14), and t(14;16)—occur in approximately 15–20%, 10–25%, and 3–7% of newly diagnosed MM patients, respectively [13]. Double translocations in MM, such as the co-occurrence of t(11;14) and t(14;16) observed in our case, are exceedingly rare. To our knowledge, this is the first reported case of t(11;14) and t(14;16) occurring together. Only one prior case report has described double translocations involving IGH, specifically t(4;14) (FGFR3/IGH fusion) and t(14;16) (IGH/MAF fusion), while eight other cases have documented double translocation myeloma with both t(8;14) (IGH/MYC) and t(11;14) (IGH/CCND1) translocations [1, 14,15,16].
In our case, all IGH loci involved in both CCND1 and MAF fusions showed no normal IGH locus (green color) detected by FISH, suggesting that the translocations originated from the same IGH loci. The IGH enhancer likely drives the high expression of both CCND1 and MAF. However, advanced molecular techniques such as optical genome mapping and DNA sequencing are needed to better understand the detailed molecular structural rearrangements underlying these double translocations.
The genetic landscape of MM can evolve over time, leading to the emergence of new chromosomal abnormalities or translocations such as t(14;16), which was not initially detected in our case. This phenomenon can be attributed to genomic instability and clonal evolution of MM, where the disease comprises multiple subclones of myeloma cells, each with distinct genetic profiles. As the disease progresses, selective pressures from the microenvironment or treatments can lead to the expansion of subclones and the acquisition of new genetic abnormalities [17, 18].
Moreover, the sensitivity of detection techniques, such as FISH and next-generation sequencing, plays a critical role. These methods may fail to detect minor subclones with specific genetic abnormalities if those subclones are present at very low levels initially but become detectable as they expand under selective pressures [19]. Furthermore, exposure to high-dose chemotherapy (e.g., melphalan) drives mutations during relapse. These mutational processes contribute significantly to the genetic complexity of MM, influencing disease progression and response to treatment [20,21,22].
Our patient’s therapeutic journey, involving multiple lines of treatment and clinical trials, highlights the challenges of managing relapsed/refractory MM with complex genetic profiles. We propose that the emergence of the t(14;16) translocation following treatment with a BCL-2 inhibitor may have contributed to disease progression by creating a selective advantage for a pre-existing t(14;16) subclone to expand or by driving its de novo emergence. In patients with t(11;14) translocation, there is overexpression of BCL-2 and usually low expression of MCL-1 and BCL-XL proteins [23, 24]. Hence, medication like venetoclax, which is a BCL-2 inhibitor, has proven to be an effective treatment [25]. The emergence of a t(14;16) translocation in a patient with a t(11;14) translocation could significantly affect the therapeutic efficacy of this medication [26]. The t(14;16) translocation leads to increased expression of anti-apoptotic proteins BCL-XL and MCL-1 [27]. This shift in the expression of anti-apoptotic proteins may have made this patient refractory to venetoclax [23, 27].







Comments (0)