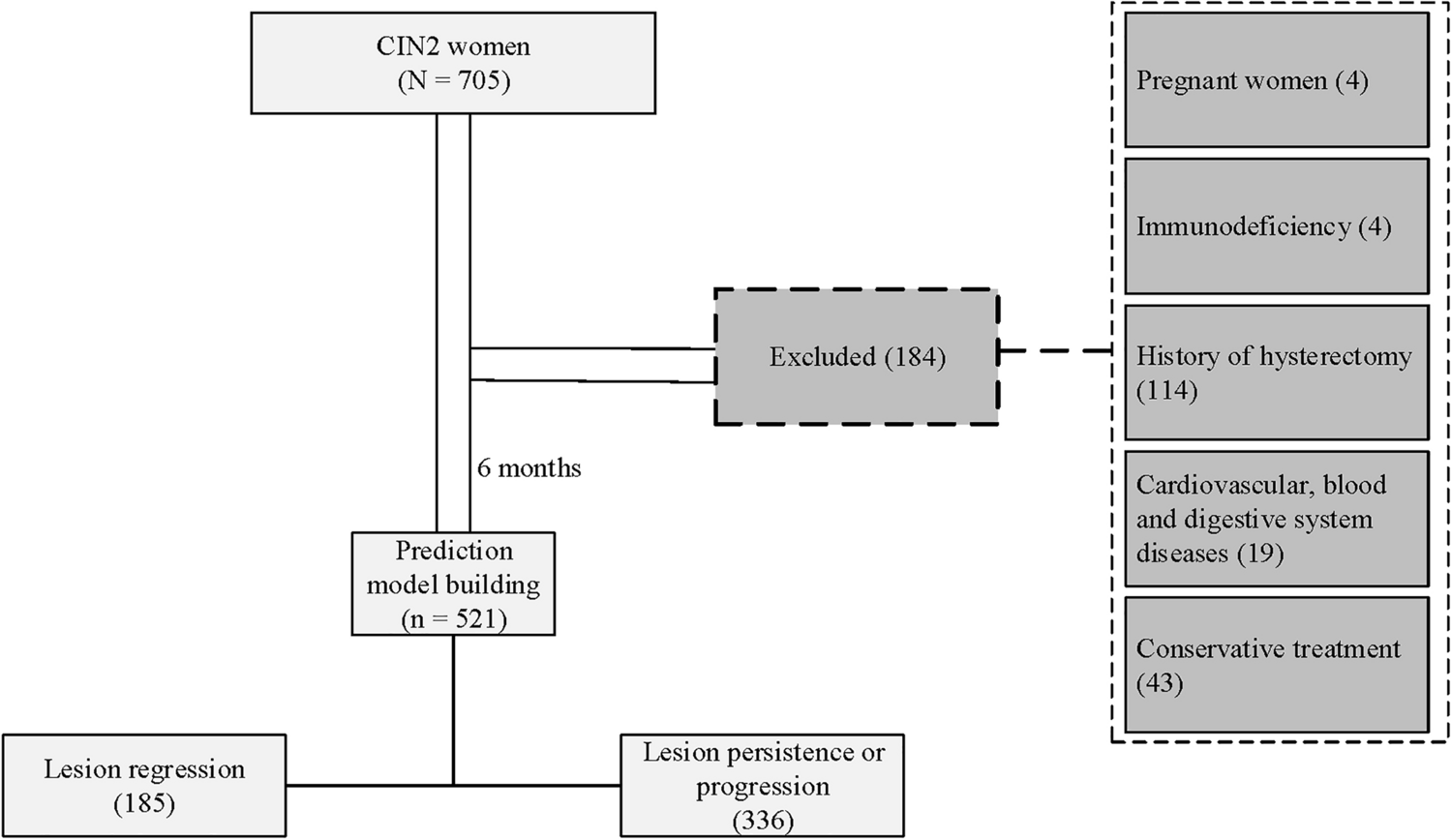
2.1 Patients and samples
This study included 120 patients with CRC treated at Henan Provincial People's Hospital from January 2015 to December 2017. All procedures involving human participants adhered to the Declaration of Helsinki (as revised in 2013), and the study design was approved by the Ethics Committee of Henan Provincial People's Hospital, China (approval number. 2022080). Informed consent was obtained from all the patients or their families. The inclusion criterion was no preoperative chemoradiation or chemotherapy. The clinical data were complete, and the postoperative pathological diagnoses were confirmed by two pathologists. Fresh tissue samples were frozen at −80 °C for western blotting experiments. A total of 120 patients with CRC were followed up until December 31, 2022.
2.2 Bioinformatics analysis
Differential expression of NEK2 in cancer (colon adenocarcinoma, COAD) and normal tissues was evaluated via The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) data in Gene Expression Profiling Interactive Analysis (GEPIA) [23]. The threshold for differential expression of NEK2 was set at |log2FC|> 1.5 and p < 0.05. The expression of NEK2 in cancer tissues of different stages was also analyzed with GEPIA (http://gepia.cancer-pku.cn/). The correlation between NEK2 expression and the OS of CRC patients was evaluated with the Kaplan–Meier plotter database (https://kmplot.com/analysis) [24]. A hallmark gene set (h.all.v7.2.symbols.gmt) was used for enrichment analysis in Gene Set Enrichment Analysis (GSEA) v 4.1.0 software (https://www.gsea-msigdb.org/gsea/index.jsp) [25].
2.3 Cell line treatment
HCT116 (CBP60028), HT29 (CBP60011), SW480 (CBP60019), and SW620 (CBP60036) cells and colorectal-mucosal cells (FHC, CRL-1831) were purchased from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The cells were authenticated through short tandem repeat (STR) profiling, and no cross-contamination was identified following mycoplasma testing. The cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (11,875,093, Gibco, Grand Island, CA, USA) containing 10% fetal bovine serum (FBS, 5,670,401, Gibco) at 37 °C in a 5% CO2 incubator. Small-interfering RNAs (siRNAs) targeting NEK2 (si-NEK2#1, 5'-AAACAUCGUUCGUUACUAU-3', si-NEK2#2, 5'-GGAUCUGGCUAGUGUAAUU-3', si-NEK2#3, 5'-GAGACUAGCAGAGGACAAA-3'), negative control siRNA (si-NC, 5'-UUCUCCGAACGUGUCACGUT T-3'), overexpression plasmids (pcDNA3.0-NEK2), and empty vector plasmids (pcDNA3.0-NC) were manufactured by GenePharma (Shanghai, China). The sequences of siRNAs (si-NEK2#1, #2, #3) and Full-length human NEK2 cDNA sequences were obtained from the National Center for Biotechnology Information (NCBI) reference sequence (NM_001204182.2). CRC cells were transfected with si-NEK2, si-NC, pcDNA3.0-NEK2 or pcDNA3.0-NC using Lipofectamine-2000 (11,668,500, Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer's instructions. The mixture was incubated for 10 min at room temperature. NEK2 was inhibited using the NEK2-specific inhibitor JH295 (4322; Tocris Bioscience, Bristol, UK). JH295 was dissolved in dimethyl sulfoxide (DMSO) and diluted with medium to a final concentration of 3 μΜ [26].
2.4 Immunohistochemistry (IHC)
Paraffin-embedded CRC tissue blocks were divided into 4 μm sections. The experimental procedure involved deparaffinization with xylene, hydration with ethanol, blocking with 4% H2O2 for 10 min, and incubation with rabbit anti-NEK2 (1:250, PAH562Ra01, CLOUD-CLONE, Wuhan, China) and rabbit anti-β-catenin (1:400, PAB021Ra01, CLOUD-CLONE, Wuhan, China) antibodies. The samples were incubated with a biotinylated goat anti-rabbit secondary antibody (1:500, SAA544Rb17, CLOUD-CLONE, Wuhan, China) for 1 h, and freshly prepared 3,3′-diaminobenzidine tetrahydrochloride (DAB, P0202, Beyotime, Shanghai, China) solution was added to develop color. After being stained with hematoxylin (C0107; Beyotime), the slides were sealed with coverslips for observation. The IHC staining results were determined on the basis of previous studies [19]. Briefly, positive cells were assessed as follows: no positive cells were scored as 0, 1%-25% 1, 26%–50% 2, 51%-75% 3, or ≥ 76% 4. The intensity of cell staining was evaluated as follows: absence of staining (−) was scored as 0, light yellow staining (+) 1, brown staining (+ +) 2, and dark brown staining (+ + +) 3. The immunoreactivity score was calculated as follows: Immunoreactivity score (IRS) = score of positive cells × score of staining intensity of cells.
2.5 Quantitative real-time PCR (qRT-PCR) analysis
Total RNA was extracted with TRIzol reagent (15596026CN, Thermo Fisher Scientific, Waltham, MA, USA). cDNA synthesis was performed using a reverse transcription kit (G3333-50; Servicebio, Wuhan, China). The PCR amplification system was prepared using 2 × SYBR Green qPCR Master Mix (G3326-01, Servicebio), and qRT-PCR was conducted on an ABI 7500 instrument (Thermo Fisher Scientific). The primers used were manufactured by Gene-Pharma (Shanghai, China). The sequences of primers used for NEK2 and GAPDH were as follows: forward: 5'-CATTGGCACAGGCTCCTAC-3', reverse: 5'-GAGCCATAGTCAAGTTCTTTCCA-3'; β-catenin: forward: 5'-GCGCCATTTTAAGCCTCTCG-3', reverse: 5'-AAATACCCTCAGGGGAACAGG-3'; glycogen synthase kinase-3β (GSK-3β): forward: 5'-TGGCTGAACTGTTGCTAGGA-3', reverse: 5'-TTTGCTCCCTTGTTGGTGTC-3'; activated protein C (APC): forward: 5'-GTGATCAGCAAACGCAGGAA-3', reverse: 5'-TGGAACTTCGCTCACAGACT-3'; and GAPDH: forward: 5'-TGGAAGGACTCATGACCACA-3', reverse: 5'-TTCAGCTCAGGGATGACCTT-3'. Gene expression was estimated using the 2−ΔΔCt method and normalized to that of GAPDH.
2.6 Cell proliferation and cytotoxicity assays
HCT116 and SW620 cells with NEK2 knockdown or overexpression were seeded into 96-well plates (5 × 104 cells/mL) for the Cell Counting Kit‑8 (CCK-8) assay (CK04, Dojindo Molecular Technologies, Kumamoto, Japan). The cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (27,016,021, Gibco) supplemented with methyl 3-(4-methylphenylsulfonamido) benzoate (MSAB, SML1726, Sigma-Aldrich, St. Louis, MO, USA) or without MSAB. Ten microliters of CCK-8 solution were added to each well, and the absorbance at 450 nm was measured via a spectrophotometer (Multiscan MK3, Thermo Fisher Scientific). RPMI 1640 medium was supplemented with 5-FU (F6627, Sigma-Aldrich) at doses of 0.5, 1, 5, 10, and 20 µg/mL for 48 h [27, 28]. The half-maximal inhibitory concentration (IC50) of 5-FU against CRC was determined. Cell viability was quantified as follows: Cell viability (%) = (absorbance of treated cells − absorbance of blank)/(absorbance of control cells − absorbance of blank) × 100%.
2.7 Colony formation assay
CRC cells (500 μL, 1 × 103 cells/mL) were added to 6-well plates and cultured continuously when visible to the naked eye. The cells were stained with 0.5% crystal violet (G1064, Solarbio, Beijing, China) for 30 min, and photomicrographs were captured via a microscope (CX43, Olympus, Tokyo, Japan). The number of colonies was quantified via Image J software (version 1.8.0; National Institutes of Health, Bethesda, MD, USA).
2.8 Transwell assay
Transwell chambers (8 µm × 3422, Corning, Inc., Corning, NY, USA) were used to assess the invasion and migration capabilities of the cells. CRC cells (1 × 104 cells/mL) transfected for 48 h were resuspended in serum-free RPMI 1640. The upper chamber was filled with 200 μL of this suspension, while the lower chamber contained 500 μL of high-nutrient solution. After 48 h of incubation, the migrated cells were fixed in 4% formaldehyde (P6148; Sigma-Aldrich) for 30 min and stained with 0.2% crystal violet (G1064, Solarbio). For the invasion experiments, Matrigel (354,234, Corning, Inc., USA) was prediluted and applied to the upper chamber before the invasion assays were conducted. Photomicrographs were captured via a microscope (CX43, Olympus). The number of cells was quantified via Image J software (version 1.8.0; National Institutes of Health).
2.9 Wound healing experiment
CRC cells were seeded onto the plates. When the cells migrated to the 90% convergence point, they were scratched. The cells were then incubated in serum-free medium for 48 h, and images of the cells that migrated to the wound area were captured via a microscope (CX43, Olympus). Image J (version 1.8.0, National Institutes of Health) was used to analyze the wound areas. Wound healing was quantified as follows: Wound healing rate (%) = (scratch area at 0 h–scratch area at 48 h)/scratch area at 0 h × 100.
2.10 Flow cytometry analysis of apoptosis
Apoptosis of HCT116 and SW620 cells was assessed via flow cytometry coupled with an Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit (C1062M, Beyotime). CRC cells (5 × 105 cells/ml) were transfected for 48 h in culture flasks. After 48 h of culture in RPMI 1640 medium, the cells were collected and centrifuged. Following fixation, the cells were washed, stained with 5 μL of Annexin V-fluorescein isothiocyanate (V-FITC) and 10 μL of propidium iodide (PI), and analyzed for apoptosis using an FC500MCL/MPL flow cytometer (Beckman, Brea, CA, USA).
2.11 Caspase-3/7 activity assay
CRC cells (5 × 104 cells/ml) were transfected for 48 h. After 48 h of culture in RPMI 1640 medium with or without 10 µg/mL 5-FU, caspase-3/7 activity was measured via a caspase 3/7 activity assay kit (MA0329; Meilunbio, Dalian, China). The fluorescence intensity was analyzed at 405 nm using a spectrophotometer (Multiscan MK3, Thermo Fisher Scientific).
2.12 Western blot analysis
Total protein was extracted from the cell lines using radioimmunoprecipitation assay (RIPA) buffer (P0013D, Beyotime). The protein concentration was subsequently quantified via a bicinchoninic acid (BCA) assay kit (P0009, Beyotime). The extracted proteins (50 μg) were separated via sodium dodecyl sulfate‒polyacrylamide gel electrophoresis (SDS‒PAGE, P0012A, Beyotime) and transferred to polyvinylidene difluoride (PVDF) membranes (YA1701, Solarbio). The membranes were incubated overnight at 37 °C with primary antibodies. Rabbit anti-NEK2 (1:400, PAH562Ra01) and rabbit anti-β-catenin (1:500, PAB021Ra01), anti-c-myc (1:400, PAB290Ra01), anti-cyclin D1 (1:400, PAA585Ra01), and anti-GAPDH (1:1000, CAB932Mi01) antibodies were purchased from CLOUD-CLONE (Wuhan, China). Rabbit anti-E-cadherin (1:800, ab212059), anti-N-cadherin (1:800, ab245117), anti-APC (1:1000, ab40778), and anti-GSK-3β (1:1000, ab227208) antibodies were purchased from Abcam (Cambridge, MA, USA). Horseradish peroxidase-conjugated goat anti-rabbit IgG (1:2000, ab205718, Abcam) was added and incubated at 37 °C for 1 h. The signals were visualized using enhanced chemiluminescence (ECL) reagent (36208ES60, Sigma-Aldrich) on an imaging system (5200 Multi, Tanon, Shanghai, China). The grayscale value was calculated via Image J software (version 1.8.0, National Institutes of Health).
2.13 Immunofluorescence assay
SW620 cells were divided into the pcDNA3.0-NEK2 group and the pcDNA3.0-NC group. The cells (1 × 104 cells/ml) were cultured on 1 cm glass slides. After fixation with 4% paraformaldehyde (100,496; Sigma-Aldrich) at room temperature, the cells were permeabilized and blocked with 0.5% Triton X-100 (83,443; Sigma-Aldrich). The cells were subsequently incubated with an anti-β-catenin antibody (1:100, 17,565–1-AP, Proteintech) and secondary antibodies (1:200, RGAR004, Proteintech). Nuclei were treated with 4,6-diamino-2-phenyl indole (DAPI, D9542, Sigma-Aldrich). Images were acquired via a fluorescence microscope (BX53, Olympus). Image J software (version 1.8.0, National Institutes of Health) was used to circle the nuclear area and analyze the mean fluorescence intensity in a blinded fashion [29, 30].
2.14 Statistical analysis
Statistical analysis and graphing were performed using SPSS-17.0 (IBM Corporation, Chicago, Illinois, USA) and GraphPad Prism-8.0 software (GraphPad Software, Inc., La Jolla, CA, USA), respectively. The quantitative data are expressed as the means ± standard deviations. A t test (two groups) was used for two-group comparisons, and one-way analysis of variance (ANOVA) followed by the post hoc Tukey test was used for multiple-group comparisons. Pairwise comparisons were performed with the (LSD)-t test. The associations between NEK2 protein expression and clinicopathological features were analyzed via rank-sum analysis. Furthermore, Kaplan–Meier analysis and Cox regression analysis were used to evaluate the prognostic significance of NEK2. In the Cox regression analysis, variables with statistical significance in the univariable Cox regression analysis were further included in the multivariable Cox model. The threshold for statistical significance was set at p < 0.05.






Comments (0)