
Remember me
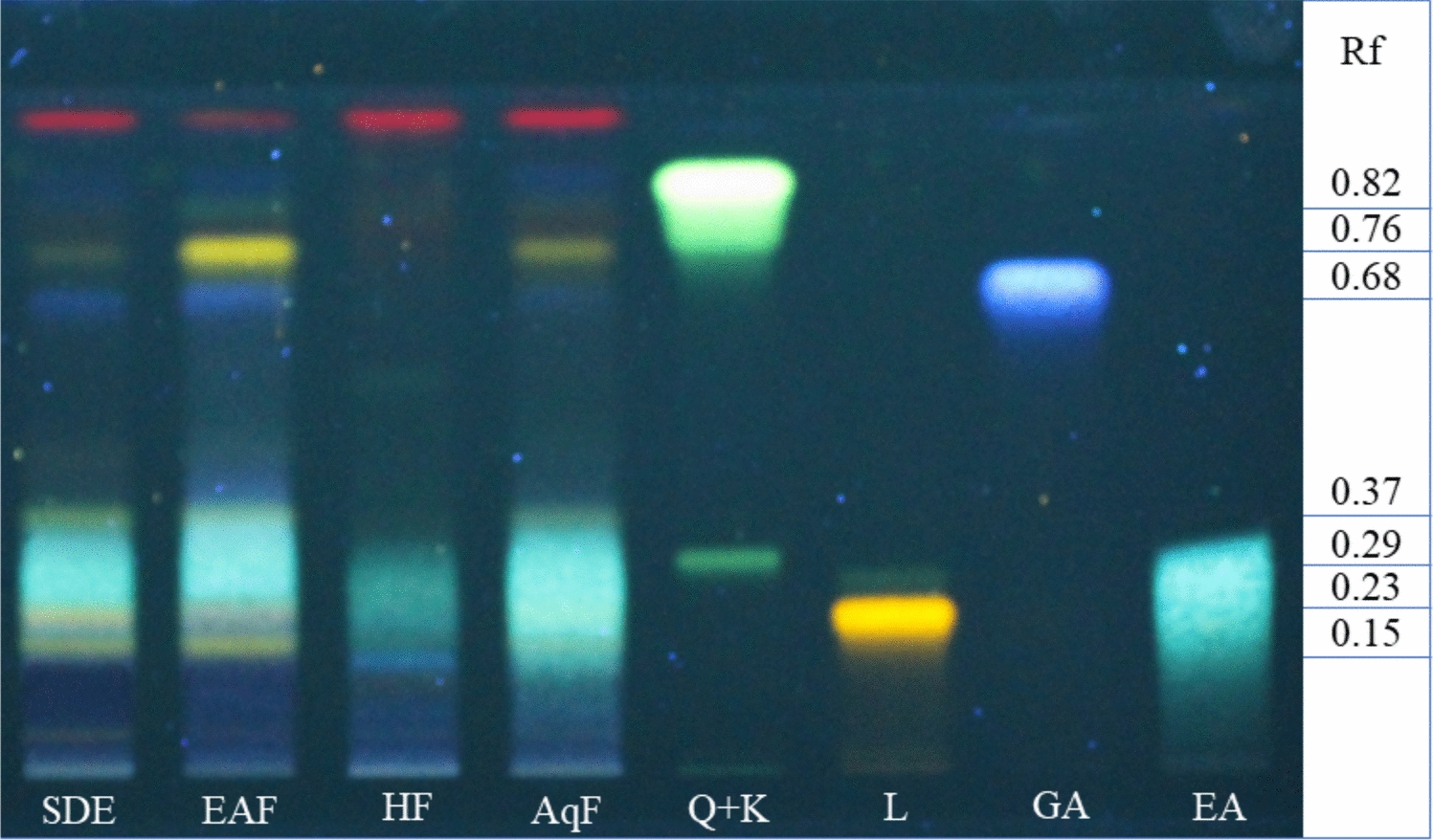





Figure 3 presents a diagrammatic representation of AD therapy based on disease severity.
Figure 3
A diagrammatic representation of AD therapy based on the severity of the condition
Topical treatmentsThe European Task Force on Atopic Dermatitis (ETFAD) policy document recommends moisturizers for all AD patients (Vestergaard et al. 2024). Topical treatment is important for AD patients with BSA below 10%. Topical therapies include topical corticosteroids (TCS) and calcineurin inhibitors (TCI). Over 40 novel topical chemicals are being developed in a Phase 3 clinical study. Emerging medicines target the aryl hydrocarbon receptor (AhR), Janus kinase (JAK-receptors (licensed in the US as Ruxolitinib and in Japan as Delgocitinib), TRPV1, and phosphodiesterase-4 (PDE4) inhibitors (licensed in the US as Crisaborole) (Müller et al. 2024).
Topical corticosteroids (TCS)Topical corticosteroids are the first-line treatment for AD, TCS reduces itching, inflammation, and redness by decreasing dendritic and Langerhans cell antigen processing and inflammatory cytokine levels (Calabrese et al. 2022). Hydrocortisone, triamcinolone, and betamethasone are commonly used for AD management. These drugs are used in creams, ointments, and lotions for ease of application. Hydrocortisone (1%) is the most effective for the face and neck, and clobetasol is the most effective for other skin areas (Kakkar et al. 2023). Although highly effective in treating AD, topical steroids can cause skin shrinkage, skin thinning, telangiectasia, and severe withdrawal when stopped. Secondary infections are also possible due to weakened immunity (Marshall et al. 2024). Children’s body surface area to weight ratio is higher than that of adults; hence, they experience a higher drug absorption rate. Indeed, many patients worry about the use of steroids during treatment. A recent study indicated that despite the enormous quantity of data supporting topical steroids for AD care, many patients are unwilling to use them (Marshall et al. 2024).
Topical calcineurin inhibitors (TCI)Topical calcineurin inhibitors (TCI) are second-line and superior alternative approaches for AD management. TCIs are costly immunomodulators and are restricted to children aged > 2 years. There are two types of TCI: Elidel® cream (pimecrolimus) at 1% for mild to moderate eczema and Protopic® ointment (tacrolimus) at 0.1 or 0.3% for moderate to severe eczema. Calcineurin inhibitors suppress IL-2 synthesis, reducing T cell activation and immunological response (Abędź and Pawliczak 2019). TCI is usually applied twice daily in tender areas, such as the face and skin folds, where corticosteroids may cause skin atrophy or thinning (Eichenfield et al. 2014).
Systemic treatmentsAlthough AD itch is mainly non-histamine, first- and second-generation antihistamines may be recommended for patients with trouble sleeping or scratching. Oral second-generation antihistamines, such as cetirizine or levocetirizine, are recommended for patients at work or school because first-generation antihistamines, such as diphenhydramine and hydroxyzine, are sedatives. Systemic immunosuppressive drugs such as, cyclosporine, azathioprine, and methotrexate are, administered when standard therapies fail. These medications are prescribed for treatment-resistant or severe AD. Cyclosporine is rather selective because it stops calcineurin from activating the nuclear factor of activated T cells (NFAT) (Vestergaard et al. 2024). Pretreatment renal and hepatic screening is recommended for systemic therapy candidates because these drugs can cause kidney or liver impairment (Kakkar et al. 2023).
New specific treatment approachesProgress on new specific treatments has grown over the past few years (Table 3). The next section discusses these novel treatments.
Table 3 Recent biological and small molecules immunomodulators for AD therapyBiologicalsComprehending the immune and genetic aspects of AD has aided identify targets for innovative treatments that more effectively target the disease’s underlying causes. Research and clinical trials have shown the practical value of biological therapy and have supported the development of patient-specific, targeted AD medicines (Caffarelli et al. 2023).
Dupilumab (Dupixent®) is an innovative US Food and Drug Administration (FDA)- and European Commission-approved therapy for children with moderate-to-severe AD aged 1 year and weighing at least 15 kg (D’Ippolito and Pisano 2018). It is an inhibitor of interleukin-4 (IL-4) receptor-α. This monoclonal antibody inhibits Janus kinase/signal transducers and activators of the transcription (JAK-STAT) signalling pathway by targeting the IL-4 and IL-13 signaling pathways. These cytokines are significant in the pathogenesis of atopic diseases. Dupilumab is usually injected subcutaneously every two weeks and is frequently used in combination with other treatments (D’Ippolito and Pisano 2018). Conjunctivitis is an adverse effect of dupilumab, and 28% of patients reported experiencing burning and a sensation of a foreign body in their eyes (Aszodi et al. 2019). SOLO 1 and SOLO 2 clinical studies involved enrolling individuals with moderate-to-severe atopic dermatitis whose condition was not well controlled by topical therapy in two randomized, placebo-controlled, phase 3 studies of the same design (Simpson et al. 2016). Patients were randomly randomized in a 1:1:1 ratio to receive subcutaneous dupilumab (300 mg) or placebo weekly or the same dose every other week alternating with placebo for 16 weeks. The primary outcome was the proportion of patients with a score of 0 or 1 (clear or almost clear) on the Investigator’s Global Assessment and a 2-point or more drop from baseline at week 16. SOLO 1 enrolled 671 patients and SOLO 2 708. In SOLO 1, the primary objective was achieved by individuals who got dupilumab every other week or weekly, compared to those who received placebo (P < 0.001). Similar results were observed in SOLO 2, with 36% of patients receiving dupilumab every other week or weekly experiencing the primary endpoint, compared to 8% of patients receiving placebo (P < 0.001). In the two trials, dupilumab significantly improved eczema area and severity index scores by at least 75% from baseline to week 16 compared to placebo (P < 0.001). Dupilumab also improved pruritus, anxiety, depression, and quality of life, pruritus, anxiety, and sadness. Dupilumab caused greater injection-site reactions and conjunctivitis than placebo.
To evaluate dupilumab’s long-term safety and effectiveness in AD patients. Adults who had previously taken part in phase 1 through 3 clinical trials of dupilumab for AD were assessed for long-term treatment in this multicenter, open-label extension study (NCT01949311). At the data cutoff (April 2016), this analysis looked at individuals who received 300 mg of dupilumab per week for up to 76 weeks. The main goal was safety, although efficacy was also assessed. The findings indicated that At cutoff, 92.9% of the 1491 enrolled patients (1042.9 patient-years) were undergoing therapy. There were no new safety signals, and the safety profile was in line with previously published trials (420.4 adverse events/100 patient-years and 8.5 serious adverse events/100 patient-years). Nasopharyngitis, conjunctivitis, and injection-site responses were among the most frequent adverse events. All efficacy endpoints, including measures of skin inflammation, pruritus, and quality of life, showed sustained improvement for up to 76 weeks. The study’s limitations include the absence of a control group, a small number of patients who received treatment for 76 weeks or more (median follow-up, 24 weeks), and patients who did not receive the recommended dosage schedule of 300 mg every two weeks. The study concluded that dupilumab’s use as a continuous, long-term therapy for patients with moderate to severe AD is supported by the safety and efficacy profile of this trial (Deleuran et al. 2020).
Health Canada, the FDA, and the European Commission has recently approved the use of talokinumab for patients with moderate-to-severe AD. This monoclonal antibody (IgG4λ) anti-IL-13, is fully humanized and competitively inhibits IL-13’s ability to bind to both the IL-13Rα1 and IL-13Rα2 receptor chains (Lytvyn and Gooderham 2023). With significant promise for efficacy and low toxicity, IL-13 downregulation is a prospective therapeutic target for AD (Lytvyn and Gooderham 2023). Patients with moderate-to-severe AD who have not responded to topical therapies are eligible for tralokinumab treatment provided they are 18 years of age or older. (Lytvyn and Gooderham 2023). For patients with moderate-to-severe AD who needed systemic medication, the ECZTRA 3 research planned a phase III trial to assess the safety and efficacy of tralokinumab in conjunction with TCS. Using a double-blind approach, patients were randomized in a 2:1 ratio to receive 300 mg of SC tralokinumab or a placebo every two weeks, plus a topical corticosteroid, if necessary, for 16 weeks. The percentage of patients who received tralokinumab at the end of the 16-week treatment period who had an IGA score of 0 or 1 and an EASI 75 was significantly higher in this experiment than in those who received a placebo. A sample of patients from both study arms was rerandomized to receive subcutaneous tralokinumab q2w or q4w for an extra 16 weeks after the initial treatment period. The majority of patients continued to respond favorably to treatment. When given tralokinumab q2w, 89.6% of patients maintained their IGA responses after 32 weeks. Furthermore, EASI 75 responses were maintained at 32 weeks in 92.5% of patients who took tralokinumab q2w. At 32 weeks, the IGA and EASI 75 responses were maintained in 77.6% and 90.8% of patients who took tralokinumab q4w, respectively (Ewulu et al. 2023).
A phase III trial called ECZTRA 7 assessed the safety and efficacy of tralokinumab plus TCS in persons with severe AD who were either contraindicated for cyclosporin A treatment or had previously experienced a failure with cyclosporin A treatment For 26 weeks, 277 adults in this multicenter, randomized, double-blind research got injections of either tralokinumab or a placebo every two weeks, in addition to TCS as needed. By week 16, more patients treated with tralokinumab plus TCS than those treated with placebo plus TCS had achieved the primary efficacy target of EASI 75. According to this trial, in persons with severe AD who are not responding to CyA therapy, tralokinumab plus TCS administered as needed significantly improves AD signs and symptoms. (Gutermuth et al. 2022).
The ECZTRA 5 trial was a randomized, double-blind, placebo-controlled phase II research that examined how tralokinumab affected vaccine immune response. In a 16-week study, 107 adult patients received tralokinumab 300 mg and 108 received a placebo q2w. At week 12, all patients received the Tdap and meningococcal vaccines, and the primary objectives were positive immunological responses to either vaccine at weeks 12 and 16. At week 16, the tralokinumab and placebo groups had similar immune responses to the Tdap (91.9% vs. 96.1%) and meningococcal (86.0% vs. 84.2%) vaccinations. This suggests that tralokinumab-treated persons with moderate-to-severe AD can continue treatment without interruption because routine non-live vaccinations are safe and effective (Merola et al. 2021). All reported studies showed no life-threatening AEs for tralokinumab. Upper respiratory tract infection, headache, somnolence, toothache, conjunctivitis, pruritus, pyrexia, nasopharyngitis, and acne were the most common systemic AEs. A localized injection site reaction with pain and erythema was described. The FDA and EMA have previously approved (2021) tralokinumab for the treatment of AD in adults; however, it is not currently authorized for use in adolescents.
Clinical trials have also examined the IL-31 monoclonal antibody nemolizumab. Among people with EASI ≥ 16 at baseline, nemolizumab therapy improved inflammation, pruritus, and sleep in a post hoc analysis of a phase 2b trial of moderate-to-severe AD (Silverberg et al. 2021). A 16-week, double-blind, phase 3 trial found that nemolizumab reduced AD pruritus more than placebo plus topical treatments (Kabashima et al. 2020).
Chen et al. (2019) investigated the in vivo effects of etokimab (ANB020), a monoclonal IgG antibody, in 12 patients with moderate to severe AD in a phase-2a study. At day 29, 83% of patients attained EASI-50 and 33% attained EASI-75 following a single systemic dosage of etokimab. In addition, a notable decrease in cutaneous neutrophil infiltration was observed compared with placebo. These findings suggest that suppressing IL-31 may be an additional part of AD therapy and may have a favorable impact on inflammatory responses (Chen et al. 2019). These findings led to the initiation of a Phase II study (NCT03533751).
Lebrikizumab is a humanized monoclonal antibody that binds soluble IL-13 and is highly selective in the non-receptor binding domain. lebrikizumab stimulates IL-13Rα2 to control endogenous IL-13 while inhibiting the IL-4Rα–IL-13Rα1 signaling complex (Moyle et al. 2019). A phase IIb trial examined the effectiveness, dose–response relationship, and safety of lebrikizumab monotherapy, with TCS utilized if needed, in 280 AD adults who failed topical treatment. Patients were randomized into four groups: 250 mg lebrikizumab with 125 mg q4w, 500 mg with 250 mg q4w, 500 mg at baseline and week 2 with 250 mg q2w, and placebo q2w. At week 16, all lebrikizumab groups showed a dose-dependent reduction in EASI scores compared to placebo, with significant differences seen as early as week 4. The lebrikizumab 250 mg group had higher response rates in secondary endpoints, such as IGA 0/1 EASI 50, EASI 75, and EASI 90. Placebo-treated patients needed three times more rescue TCS, started sooner and lasted longer, than lebrikizumab-treated patients (Guttman-Yassky et al. 2020). The ADore single-arm trial examined lebrikizumab’s safety and efficacy in adolescents with moderate-to-severe AD. Lebrikizumab was administered twice at 250 mg at week 1 and week 2, followed by a single injection q4w from weeks 4 to 52. Lebrikizumab improved skin clearance clinically from week 4 onward, with more patients improving over time. The study found that targeting IL-13 with lebrikizumab for moderate-to-severe AD in teenagers is promising (Paller et al. 2021). Similarly, other studies showed clinically significant improvements in all primary and secondary objectives (Blauvelt et al. 2021; Silverberg et al. 2023b). In all investigations, lebrikizumab demonstrated a good safety profile with no significant adverse events that could be fatal. Upper respiratory tract infections, nasopharyngitis, headaches, oral herpes, conjunctivitis, and coronavirus disease 2019 (COVID-19) were the most common systemic adverse events detected. There have been reports of injection site responses locally, with lebrikizumab groups experiencing frequency ranging from 1.3% to 5.7%. In November 2023, the EMA authorized lebrikizumab for use in AD; recently in 2024, the FDA has approved it.
Two more IL-13 inhibitors in the pipeline, eblasakimab and cendakimab, are presently being studied in phase II studies (NCT04800315). Eblasakimab is a monoclonal antibody that interferes with IL-13 and IL-4 signaling by targeting the IL-13Rα1 component of type 2 receptor. Eblasakimab was tested in AD patients in a phase Ib, double-blinded research. Preliminary data presented 52 participants randomized to receive eblasakimab at 200, 400, or 600 mg versus a placebo group. After 8 weeks, eblasakimab showed significant improvement over placebo. A decrease in EASI score was observed in all dose groups: 50% in 200 mg, 63% in 400 mg, and 61% in 600 mg, compared to 32% in the placebo group. Eblasakimab also increased EASI 50 and 75 responses compared to placebo. The peak PNRS score was lower in the 600 mg eblasakimab group compared to the placebo group. An analysis found that 400 and 600 mg eblasakimab dosages resulted in better clinical responses than 200 mg. At week 8, eblasakimab 600 mg significantly improved the mean percentage change in EASI, with more patients achieving EASI 50 and 75 compared to placebo. In addition, this study demonstrated that eblasakimab medication improved sleep disturbance and PNRS and patient-oriented eczema measure (POEM) ratings when compared to placebo. Pruritus, injection site erythema, and injection site edema were the most frequently reported adverse events (Veverka et al. 2024). Several other ongoing clinical trials are reported.
Humanized recombinant anti-IL-13 monoclonal antibody Cendakimab (RPC-4046). Cendakimab interacts to IL-13 at an epitope that overlaps with the IL-13Rα1 and IL-13Rα2 subunits, impeding their contact and signaling. A phase II multicenter, randomized, double-blind, parallel-group, placebo-controlled trial (NCT04800315) examined the efficacy and safety of three cendakimab dosage regimens in adults with moderate-to-severe AD. The study has four experimental groups. In group 1, participants received 720 mg of SC cendakimab weekly (qw) for 16 weeks. In group 2, subjects received SC cendakimab 720 mg q2w and placebo alternatively. Group 3 participants received SC cendakimab 360 mg q2w and a placebo weekly. Placebo subjects got SC placebo qw. The study’s main endpoint was the mean percentage change in EASI scores from baseline to week 16. In the trial, cendakimab 720 mg qw resulted in a higher drop in EASI score (-84.41%) compared to the 720 mg q2w, 360 mg q2w, and placebo groups (-76.03, 78.93, and 62.7%, respectively). In the cendakimab 360 mg q2w group, 38.2% of patients attained an IGA score of 0 or 1, compared to 33.3% in the 720 mg qw group and 24.4% in the 720 mg q2w group. The most common adverse effects in this study were upper respiratory tract infection, allergic conjunctivitis, COVID-19, and headache (Blauvelt et al. 2024). Even though cendakimab passed its phase II trial, development has stopped. After meeting the primary goal, cendakimab’s manufacturer observed that AD treatment is extremely competitive and that it does not offer a significant advantage over other medications (Teixeira et al. 2024). Table 4 presents clinical trials that investigated new biologics for the treatment of atopic dermatitis (AD)
Table 4 Summary of clinical studies on new biologics and small molecules for atopic dermatitis (AD), including their mechanisms of action, side effects, and conclusionsSmall moleculesCompared to biologics, small molecules (<0.5 kDa) intreat more frequent dosing and produce more off-target effects when administered systemically. Phosphodiesterase-4 (PDE-4) and Janus kinase (JAK) inhibitors represent new therapeutic options for targeting the underlying inflammatory processes in AD (Kamata and Tada 2023).
An innovative approach to managing inflammatory and immunological disorders is using Janus kinase inhibitors (JKI). Phosphotransferases known as Janus kinases (JK) are activated enzymatically when cytokines bind to their receptors (Berbert Ferreira et al. 2020). JAK inhibitors target Janus kinases, which are important signaling pathways that connect different cytokine receptors to STAT transcription factors and are involved in cytokine generation. Since this signaling pathway provides the basis for the expression of IL-2, IL-4, and numerous other cytokines, JAK inhibitors offer a promising treatment for AD (Ahluwalia et al. 2017). Different etiological processes, including hematopoiesis, immunological fitness, tissue repair, inflammation, apoptosis, and adipogenesis, are mediated by the JAK-STAT downstream signaling pathway and are linked to a range of diseases and pathological behaviors (Maji et al. 2024). Different JAK inhibitors have different selectivity, degree of inhibition, and safety characteristics (Milakovic and Gooderham 2021). JAKi have the potential to revolutionize the treatment of some people with AD, and their benefit–risk ratio appears to be reasonable in this patient population (Schwartz et al. 2018).
For AD in adults and children aged more than two years, Difamilast is licensed in certain countries (Saeki et al. 2022). There were significant differences between the difamilast and placebo groups (18.1%) in a 4-week phase III research in children with mild-to-moderate AD. There is an ongoing phase III trial for infants older than three months to 2 years. Phase III trials involving 0.15% roflumilast cream in adults and children ≥6 years of age have produced encouraging preliminary data. A three-year-old phase III trial is now occurring (Saeki et al. 2022).
Baricitinib is a first-in-class oral JAK1/JAK2 inhibitor approved in 2020 by the European Medicines Agency for adults ≥ 18 years for moderate-to-severe AD however, is not yet been FDA-approved for AD in the United States. In clinical trials. baricitinib was superior to the placebo group (Yim et al. 2024). In all groups, nasopharyngitis, herpes simplex, headache, and back discomfort were the most frequent adverse events (≥5%). Pediatric investigations are ongoing (Bieber et al. 2021; Yim et al. 2024).
After topical and oral adminstration, the first-generation, non-selective JAK inhibitor delgocitinib (JTE-052/LEO 242549) improved allergic contact sensitization and D-like inflammation in animal models (Yamamoto et al. 2020). After phase III studies, Japan approved topical delgocitinib ointment for moderate to severe AD in 2020 (Nakagawa et al. 2020; Chovatiya and Paller 2021). However, cream development for chronic hand eczema (LEO 24249) has been discontinued in the United States and Europe (Chovatiya and Paller 2021).
In 2021, ruxolitinib, a JAK1/JAK2 JAKi 1.5% cream, received FDA approval for the treatment of mild-to-moderate AD ≥12 years. It has not been a widely used component of the traditional therapy arsenal (Yim et al. 2024). Ruxolitinib’s 8-week efficacy in the Investigator’s Global Assessment (IGA) and itch reduction compared to the vehicle were reported in two phase 3 trials (Papp et al. 2021; Papapostolou et al. 2022). The critical investigation revealed that the treatment was effective and tolerated for itch alleviation. Nasopharyngitis is the most prevalent side effect (Kleinman et al. 2022).
Tofacitinib, a first-generation small molecule JAK1/3 inhibitor, the FDA in November 2012 approved as a 5 mg twice daily (bid) oral dosage for moderate-to-severe rheumatoid arthritis in adults. Topical Tofacitinib was tested for mild-to-moderate AD. IN A 4-week, phase 2a randomized double-blinded vehicle-controlled trial (RDBVCT) in adults 18 to 60 years old, Tofacitinib 2% ointment compared to vehicle satisfied the main endpoint of percentage change from baseline eczema area and severity index (%EASI) at week 4 (81.7% vs 29.9%) (Bissonnette et al. 2016). Tofacitinib 2% ointment achieved secondary endpoints such as improved IGA scores (IGA 0/1) and %IGA, %BSA, and %itch-NRS, detected as early as day. Nasopharyngitis and urinary tract infections were the most common treatment-emergent adverse events (Chovatiya and Paller 2021). However, topical tofacitinib is not yet commercialized.
Second-generation JAK inhibitors, such as upadacitinib and abrocitinib, have been developed in the pursuit of JAK1-selective drugs, which are crucial for the activity of multiple cytokines implicated in AD. These agents have demonstrated rapid onset of action and high efficacy in clinical trials showing superior efficacy compared to some established treatments. Both have been approved by the European Commission, Health Canada, and the FDA for use in moderate-to-severe AD patients aged 12 years and older (Lytvyn and Gooderham 2023). In particular, JAK1-selective inhibitor SHR0302 has been developed for oral use in moderate-to-severe AD and topical application in mild-to-moderate AD. (Lin et al. 2020). In a phase III study, upadacitinib (approved in 2019 for rheumatoid arthritis) and abrocitinib improved patient outcomes and pruritus severity. (Blauvelt et al. 2021). Upadacitinib has shown promising results in long-term studies. Data indicate that upadacitinib maintains its efficacy in reducing AD symptoms over prolonged use, with an acceptable benefit-risk profile. Common adverse events include acne, nasopharyngitis, and headache, with serious adverse events being infrequent. Abrocitinib has been evaluated in long-term studies, demonstrating sustained efficacy in symptom reduction. The safety profile remains consistent with short-term studies, with nausea, headache, and nasopharyngitis among the most reported adverse events. Serious adverse events are rare but necessitate monitoring. Additionally, abrocitinib reduced itching better than dupilumab (Bieber et al. 2021; Lytvyn and Gooderham 2023).
Cerdulatinib’s combined targeting of JAKs and SYK is a promising topical pharmaceutical intervention. SYK is a crucial signaling protein for the FcεRI receptor on epidermal Langerhans cells, inflammatory dendritic cell count, and T-cell activation (Klaeschen et al. 2021). It was also reported that Cerdulatinib gel reduced epidermal thickness, inflammatory dendritic cells, and transcriptome inflammatory signatures in mild to moderate AD in a phase Ib study (Kim et al. 2020). This phase 1b, single-center, double-blind trial examined the safety and efficacy of cerdulatinib gel 0.37% and its effects on lesional (LS) skin molecular profiles in individuals with mild-to-moderate AD. Mild-to-moderate AD patients (N = 10) were randomized 4:1 to twice-daily topical cerdulatinib gel 0.37% or vehicle for 14 days. In the cerdulatinib group (n = 8; female = 4), the mean age was 30.8 years, baseline mean (SD) eczema area and severity index (EASI) was 4.0 (2.0), body surface area affected was 4.3% (2.0), and pruritus numeric rating scale score was 4.4 (1.8). In the vehicle group (n = 2; female = 2), mean age was 23.5 years, baseline mean (SD) EASI was 2.4 (0.8), body surface area affected was 3.0% (0.0), and pruritus numeric rating scale score was 1.5 (2.1). Eight of 10 individuals had 35 treatment-emergent adverse events, all mild (34 of 35; 97%) or moderate (1 of 35; 3%). Cerdulatinib’s most prevalent treatment-emergent adverse event was headache, which affected 3 of 8 participants. There were no clinically significant impacts on vital signs and electrocardiograms, and no patients quit treatment due to adverse events (Piscitelli et al. 2021).
Gusacitinib selectively inhibits JAK and SYK, which are tyrosine kinases activated by proinflammatory cytokines such IL-1β and IL-17. Gusacitinib appeared to be safe and well-tolerated in an early phase 1b study (RDBPCT). Only the highest dosages of gusacitinib met the EASI-50 secondary objectives and baseline itch-NRS improvement (Bissonnette et al. 2019). A separate phase 2b study (study to evaluate ASN002 in subjects with moderate to severe chronic hand eczema; NCT03728504) showed promising efficacy and safety (Rodenbeck et al. 2016). A double-blind, placebo-controlled, multicenter, phase 2 trial was conducted to assess the safety and effectiveness of gusacitinib (Jimenez et al.
Comments (0)