The MorbidGenes Panel provides a comprehensive dataset of disease associated genes, aiding diagnostic labs with a monthly updated list of genes relevant for routine diagnostics.
Lists of diagnostically relevant genes manually curated by dedicated reviewers, like the Mendeliome panel by PanelApp Australia with currently more than 3900 green genes are in fact needed, but the huge efforts undertaken to maintain such a list are reflected by the nearly daily activity of the 69 reviewers of the panel. In addition, global efforts to share gene-disease lists by the global community like GenCC are highly commendable, but lag behind in curation and thus rather represent a 2nd level of evidence. As GenCC gathers evidence from different data sources but does not generate new evidence itself, the GenCC database is in fact not needed for the curation of the MorbidGenes panel because it does not add unique genes to the panel. Still, the displayed evidence in GenCC provides a comprehensive overview of all available gene-disease curations, and for this reason the link to the available GenCC entry was retained in the MorbidGenes panel.
The drawback of our approach here is the loss of resolution, as only the gene name and the respective sources are retained. This is in contrast to manually curated panels like PanelApp, which provide definite gene-disease associations, allelic disorders, clinical synopses, mode of inheritance and links to publications supporting this evidence. However, the MorbidGenes Panel does not aim to be another curated gold-standard, but rather serves as a first-in-line tool for a fast detection of clinically relevant genes that need to be integrated into genetic diagnostic routine. The drawback is compensated by providing links to the respective sources on the website.
Applying the MorbidGenes Panel as an in silico panel in routine diagnostics reduces the number of variants to evaluate from about 60.000 to about 20.000 per exome, eliminating variants in diagnostically irrelevant genes. We are aware of the fact that our MorbidGenes Panel still may include false positive genes, e.g. based on somatic variants classified as pathogenic in ClinVar or old and unconfirmed OMIM entries. As we wanted to establish a simple logic for inclusion of a gene and also keep manual curation to a minimum, we decided to also keep genes in the MorbidGenes panel with a Morbidscore of 1 only, which might rather be considered a gene of unknown significance (GUS). A gene may only be supported by limited evidence based on a single publication with, for example, five individuals with a de novo missense variant – but if the analyzed individual has one of the exact same variants as reported in said publication and an overlapping phenotype, this would trigger an inclusion of the variant in the genetic report and add more value to a GUS. Although evidence is limited on a number of genes, a case specific evaluation of the genotype and phenotype of the individual is always needed to decide which variants are to be reported back to the referring clinicians. As the Morbidscore represents the number of databases that support the evidence for a certain gene, the user can set an individual threshold to filter more stringently for genes with a minimal number of supporting databases.
As Fig. 1 illustrates, it is of particular importance to include a heterogeneous set of data sources in panel curation, as important genes can be missed when focusing only on evidence from single databases. This is especially true for manually curated panels, as 282 and 407 genes are exclusively curated as a green gene in the Genomics England PanelApp and the PanelApp Australia, respectively, further undermining the need of diverse datasets. Regular updates are another important pillar in panel management, as new disease causing genes emerge continuously. More than 130 morbid genes were added from January to December 2023 (Fig. 1A), each representing a potential diagnosis which might be missed if panels are not regularly updated. Phenotype-based approaches, like filtering for genes with specific HPO terms, harbours the risk of excluding relevant variants, as systematic descriptions of phenotype-gene-associations in databases like HPO lag behind and take an amount of time to be updated. It is therefore crucial in routine diagnostics not only to include genes with minimal sufficient evidence, but also to keep the diagnostic pipelines up-to-date for potential re-evaluations of diagnostic exomes (Halfmeyer et al. 2023). Both aspects can be easily realised with our MorbidGenes Panel.

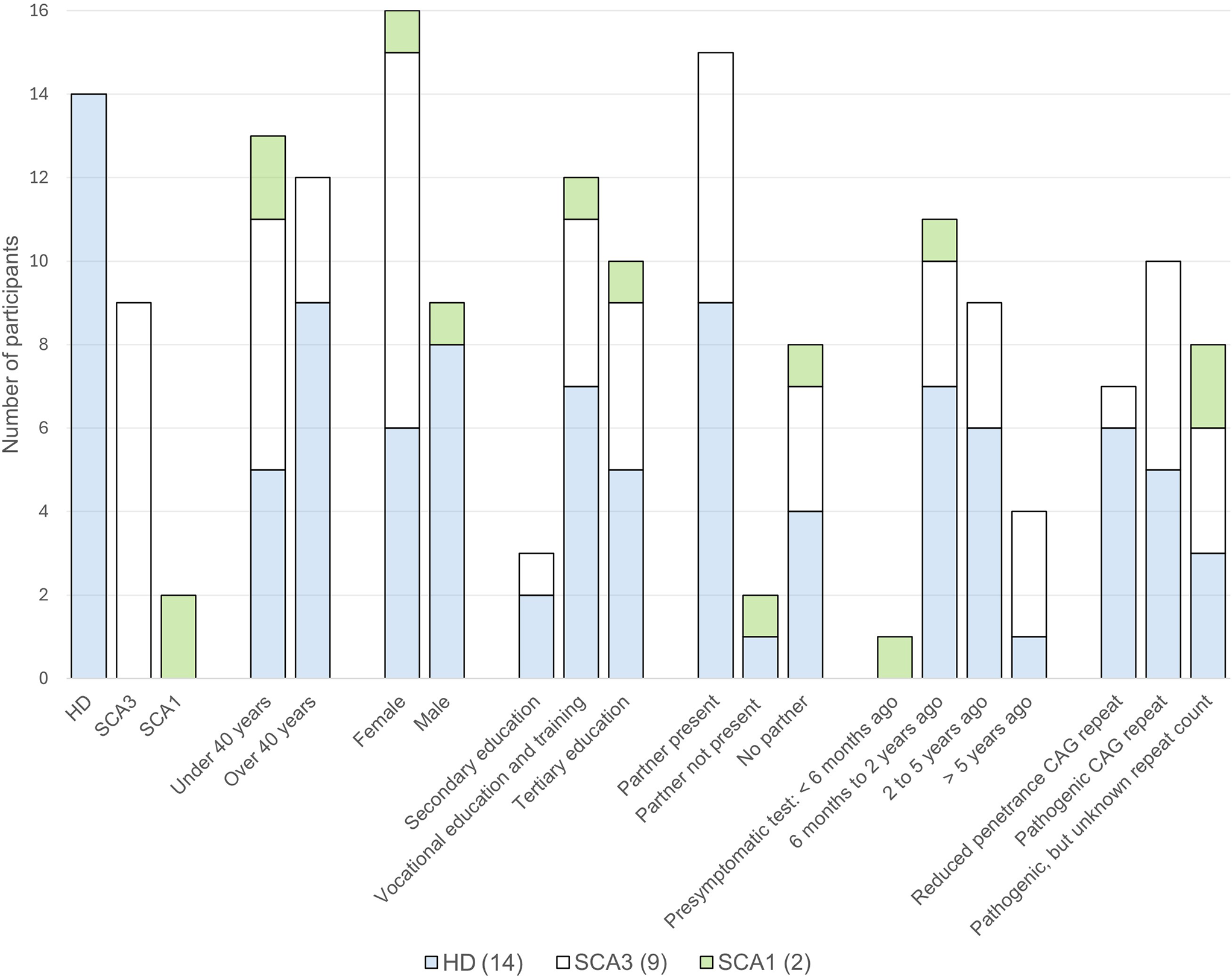

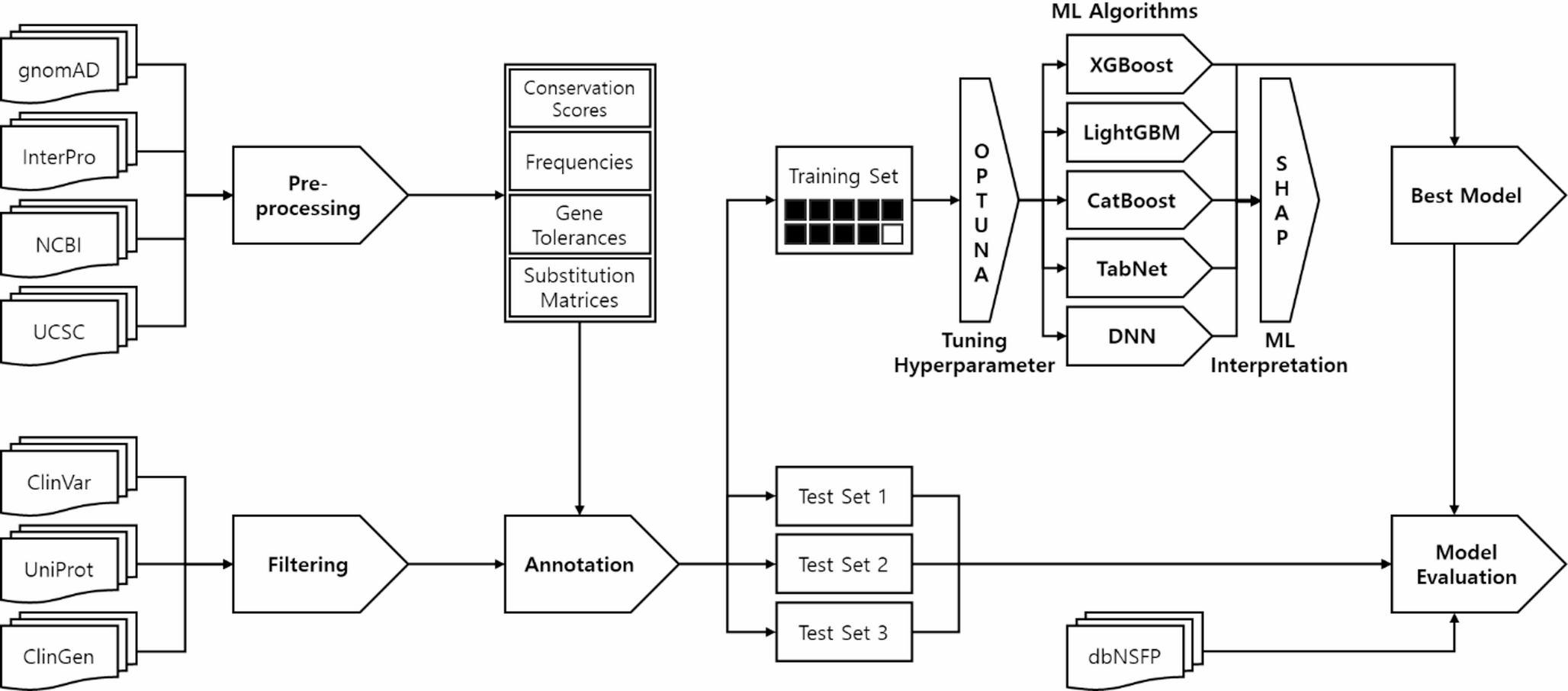
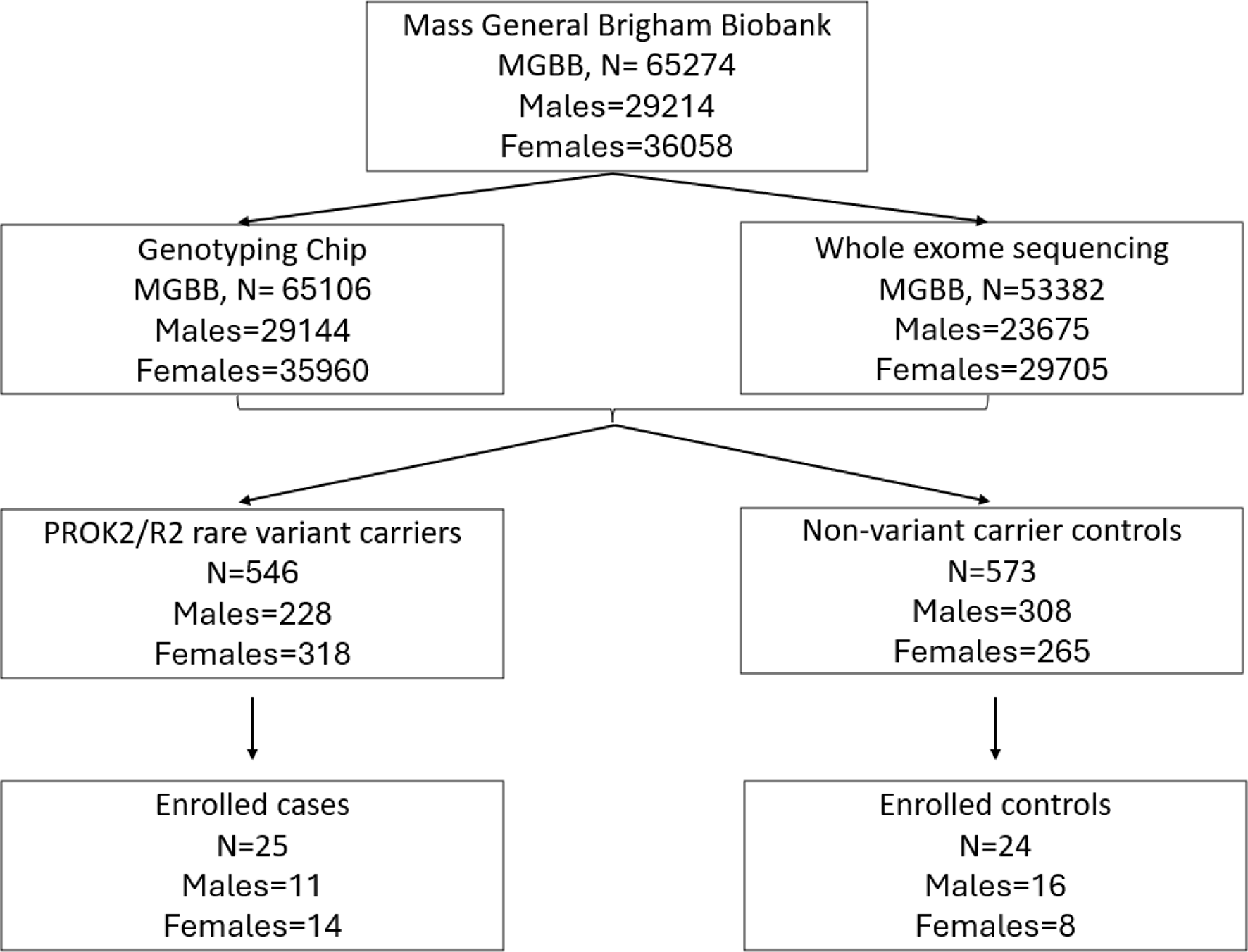
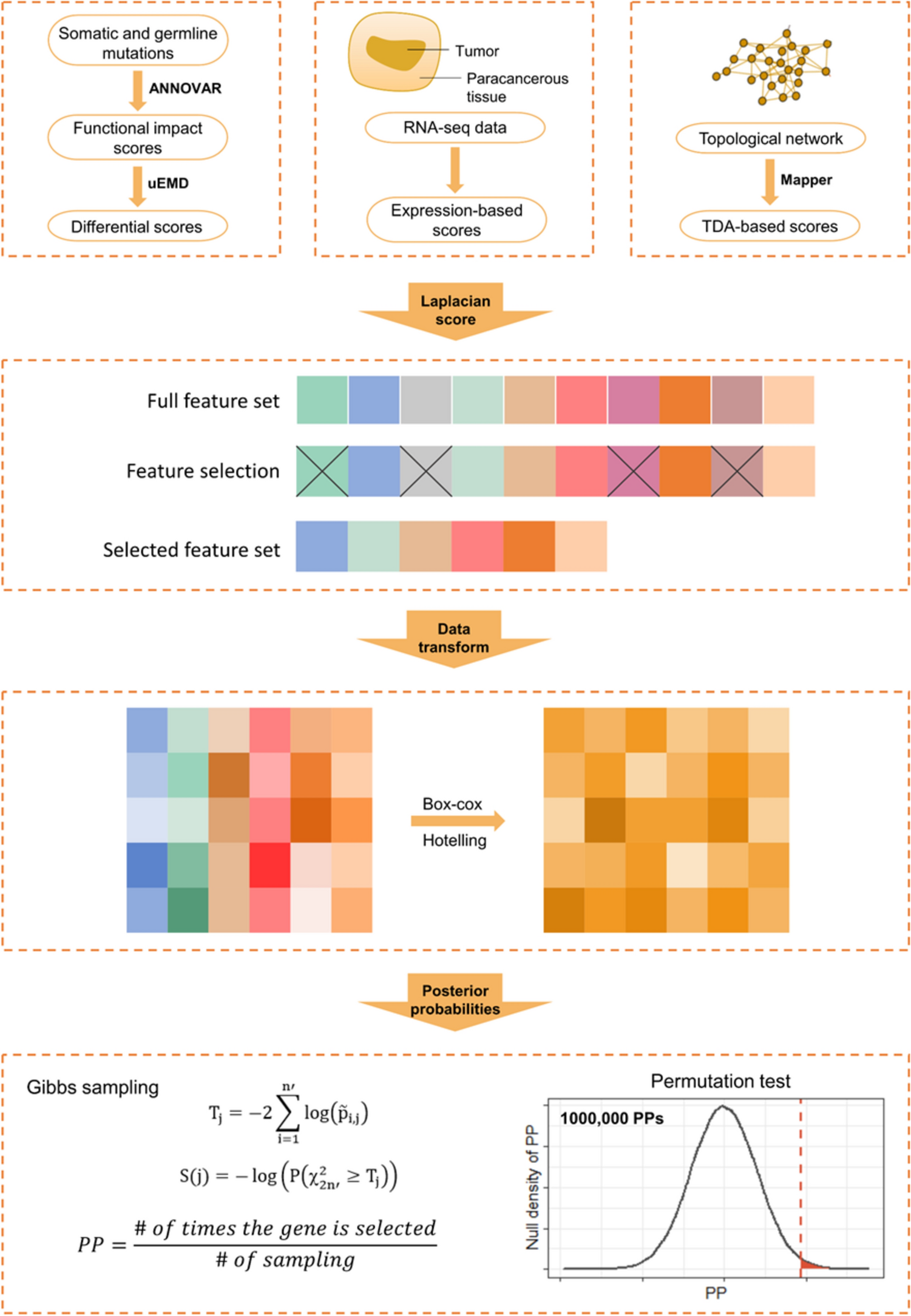
Comments (0)