
Remember me
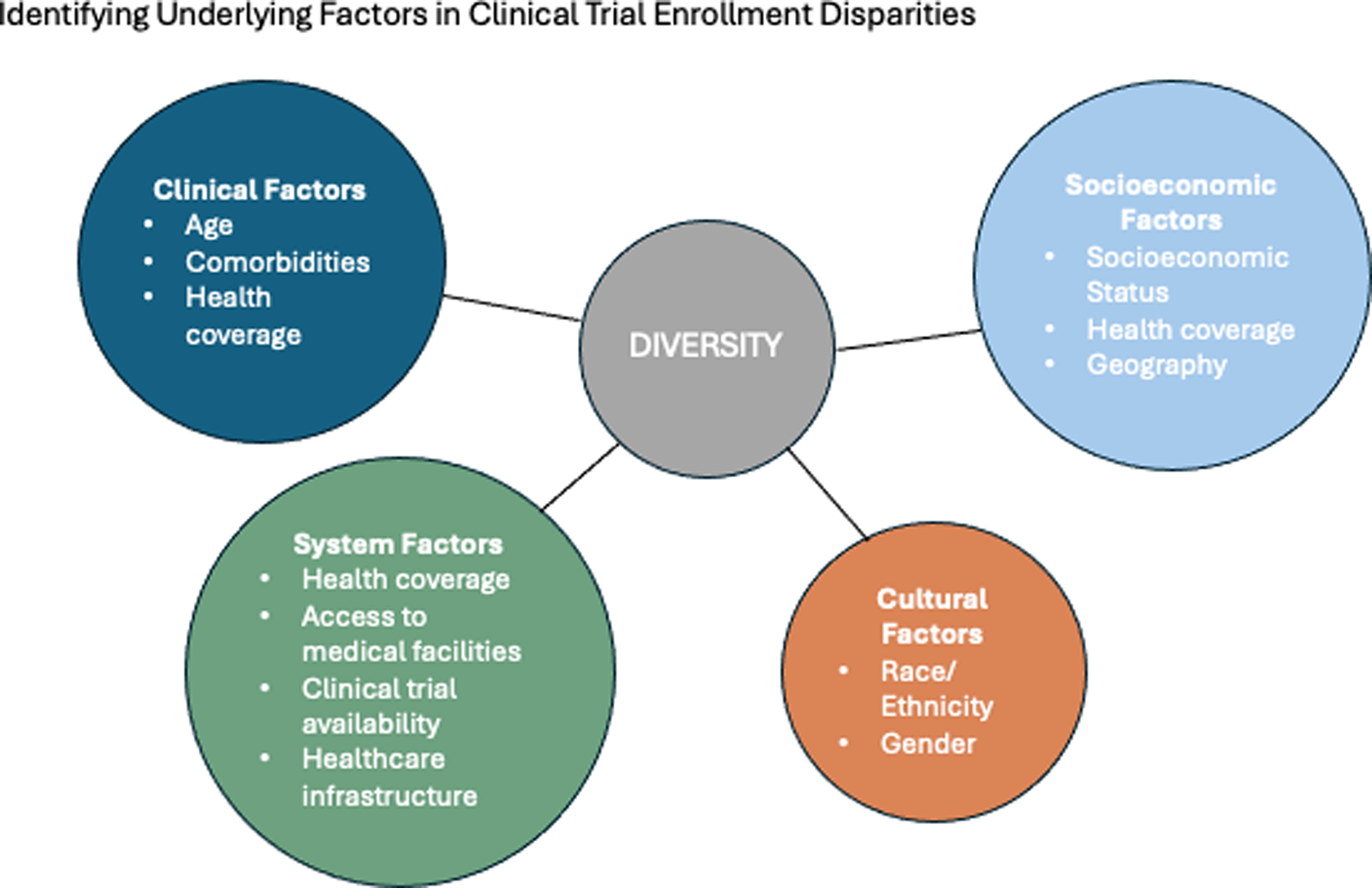



The Philadelphia chromosome negative myeloproliferative neoplasms (MPN) are a group of onco-inflammatory conditions which involve aberrant activation of JAK-STAT pathway resulting in excessive and clonal proliferation of mature myeloid blood cells, and release of abnormal cytokines [1].
Myelofibrosis (MF) is a MPN characterized by bone marrow fibrosis, dyshemopoiesis, constitutional symptoms and extramedullary hemopoiesis. It can be classified as either Primary Myelofibrosis (PMF) or secondary through progression from Polycythemia vera (Post Polycythemia myelofibrosis; PPVMF) or Essential thrombocythemia (Post essential thrombocythemia myelofibrosis; PETMF) [2].
There has been a major change in the landscape of MPNs since the discovery of the activating V617F mutation in Janus kinase 2 (JAK2) in 2005 and the subsequent development of JAK inhibitors (JAKis) [3]. However, despite these advances, the management of MF with JAKis remains challenging due to limitations in efficacy, increased toxicity and development of treatment resistance.
Pathophysiology of MyelofibrosisIn MF the most common driver mutations are in JAK2, MPL and CALR genes [4]. Generally, 65% of PMF patients carry the JAK2 V617F mutation, while 20–30% have a CALR mutation and 5% have a MPL mutation [3,4,5,6]. By contrast in those with secondary MF, JAK2 V617F is present in almost all cases of PPVMF and in 50% of PETMF, while CALR and MPL account for 30% and 10% of cases respectively [7].
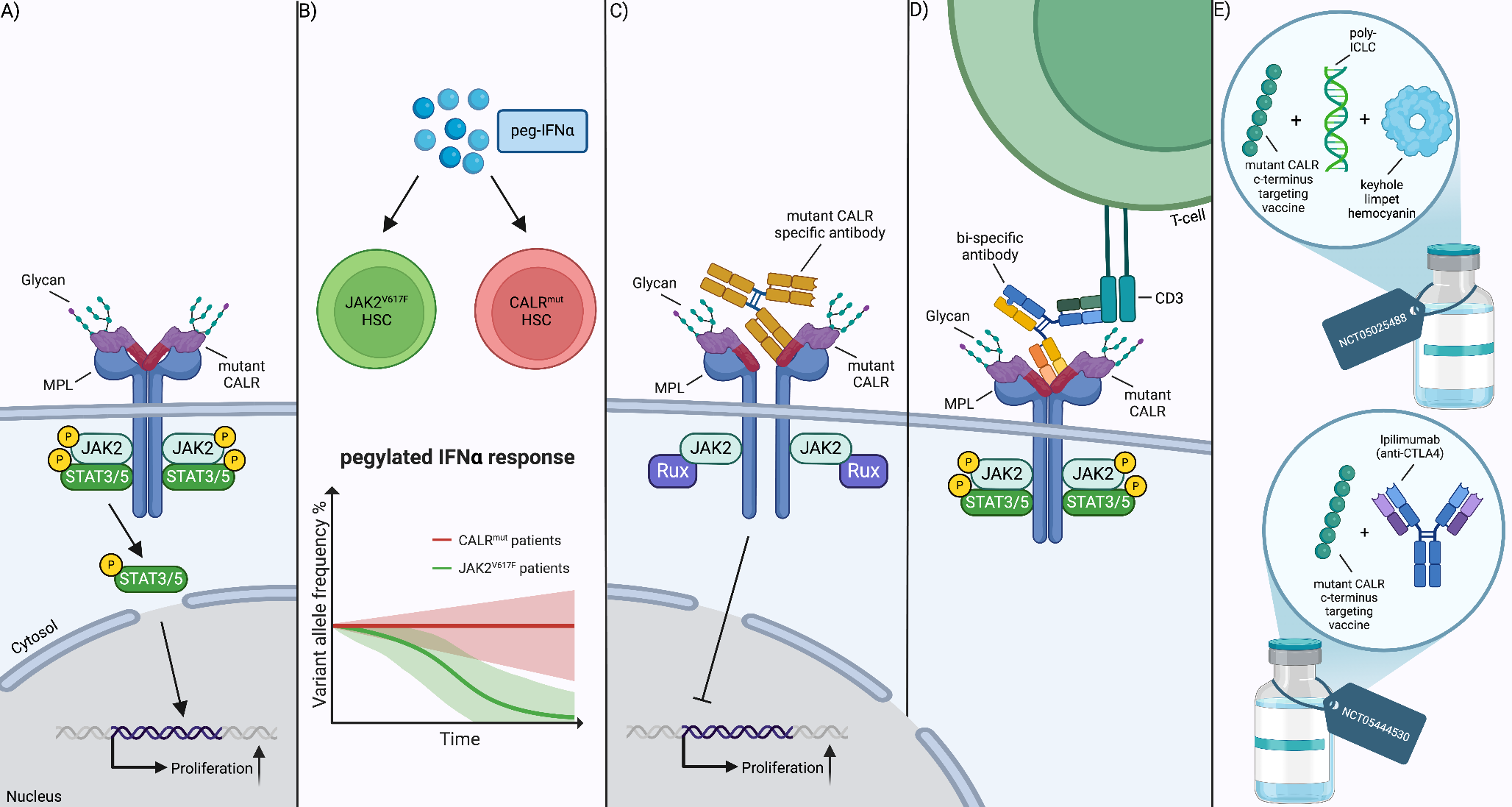
The pathophysiology for the development of this condition is through the JAK2-STAT pathway. Usually, JAK2 is activated by a variety of receptors including erythropoietin (EPOR), thrombopoietin (TPOR) and granulocyte/macrophage colony-stimulating factor (GM-CSF). Following stimulation, the JAK2-STAT pathway is activated, which then regulates the transport of transcription factors (STATs and FOXO) to the cell nucleus. In MF, JAK2-STAT signaling is constitutively activated by the driver mutations, causing excess cell proliferation, differentiation, and survival (Fig. 1) [8].
Fig. 1
JAK2-related signaling pathways as well as associated inflammatory signaling commonly associated with MF. Mutations in CALR (CALRmut), MPL (TPORW515) and JAK2 (JAK2V617F) lead to the constitutive activation of JAK2-STAT as well as PI3K/AKT and MAPK/ERK which promote the transport of transcription factors (FOXO/STATs) to the nucleus. JAK2 also activated the Toll-Like receptor (TLR) pathway which affects the production of TNF-α and reduces its sensitivity to the anti-inflammatory cytokine IL-10. MPNs also secrete high levels of pro-inflammatory cytokines, including IL-1, which activates NFκB pathway. This in turn produces high levels of IL-8 which stimulates STAT3 and PI3K/AKT thus driving cell proliferation. Overall this highlights the cross-talk between various signaling pathways in myeloproliferative neoplasms
However, JAK2 also activates other major signaling pathways, including MAPK/ERK and PI3K/AKT. In the former, ERK is a key regulator of the cell cycle as well as multiple transcription factors while in the latter AKT inhibits apoptosis and activates translation via mTOR (Fig. 1) [8, 9]. Therefore, these pathways also act as drivers in MF.
As well as the above, JAK2 activates non-canonical signaling pathways including toll-like receptor (TLR) pathway which affects production of the inflammatory cytokine TNF-α (Fig. 1). In MF, JAK2 V617F causes a defective negative regulation of TLR signaling resulting in the increased production of TNF-α and reduced sensitivity to the anti-inflammatory cytokine IL-10 [10].
Chronic inflammation is a characteristic feature of MPNs, including MF, and levels of inflammatory cytokines are typically increased in MPN patients [11]. Notably, JAK-STAT signaling is not the only contributor, as JAK2 inhibition has not been sufficient to normalize cytokine levels. The NFκB signaling pathway has since also been implicated in driving inflammation in these patients (Fig. 1) [12].
Overall, the pathophysiology of MF is far more complex than previously anticipated, with multiple signaling pathways implicated. Therefore, although JAK inhibition may be clinically effective, there is an increased risk of adverse events due to potential off-target effects. Importantly, by targeting specifically JAK2-STAT signaling, parallel pathways can potentially become overactivated thus driving drug resistance.
Diagnosis and Risk Stratification of MyelofibrosisThe diagnosis and classification of MF has recently been updated and published by both the World Health Organization (WHO) and the International Consensus Classification (ICC), with the latter summarized in Table 1. In general, the 2022 WHO classification remains mostly unchanged compared with the fourth edition [2, 13] with only a minor change from palpable to splenomegaly detected on imaging. As noted in Table 1, those patients with PMF, the Dynamic International Prognostic Scoring System (DIPSS) as well as the DIPSS Plus (DIPSS +) incorporate clinical and cytogenetic features to risk stratify patients at any timepoint during the disease course [14, 15]. In secondary MF, a separate score was developed by Passamonti et al. [16] which was validated in 685 patients diagnosed with PPVMF and PETMF known as Myelofibrosis Secondary to PV and ET-Prognostic Model (MYSEC-PM) and is used in current clinical practice (Table 1) [16].
Table 1 ICC 2022 diagnostic criteria for both primary and secondary myelofibrosis as well as the prognostic models for risk stratification [13,14,15,16, 18,19,20]In 2013, Vannucchi et al. discovered that ASXL1, SRSF2 and EZH2 mutations inter-independently predicted shorted survival [17]. This was later supported by Tefferi et al. which demonstrated that more than 80% of PMF patients harbored mutations in other myeloid genes including ASXL1, TET2, EZH2, SFSF2, DNMT3A, U2AF1 and IDH1/2 [18]. These mutations impacted overall and leukemia-free survival, independent of both the DIPSS + score as well as JAK2/CALR/MPL mutation status. Subsequently, prognostic scores were developed which incorporated these mutations and are summarized in Table 1 [19,20,21].
The main objective of these models are to facilitate treatment decisions, especially in MF patients eligible for allogeneic hematopoietic stem cell transplant (AHSCT). However, despite AHSCT being the only curative option, the morbidity and mortality risk remains high [22, 23]. Therefore, JAKis still have a major role in the treatment of MF.
Management of MyelofibrosisBased on the current National Comprehensive Cancer Network (NCCN) guidelines, management of MF is dependent on risk stratification as well as symptom burden [24]. The latter is usually assessed using the Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score (MPN-SAF TSS) which is calculated as the mean score for 10 items including fatigue, bone pain and pruritus [25].
Overall, those patients with lower-risk MF (i.e. DIPSS ≤ 2; DIPSS + ≤ 1; MYSEC-PM < 14) are managed according to their symptoms. If asymptomatic, the general recommendation is to observe, while those who are symptomatic should be commenced on cytoreductive therapy. Recommended options include Hydroxycarbamide, Pegylated Interferon alfa-2a or JAKis [24].
In those with higher-risk MF (i.e. DIPSS > 2; DIPSS + > 1; MYSEC-PM ≥ 14), treatment is dependent on the platelet count and whether they are transplant-eligible or not. If patients are not transplant-eligible, or transplant is not feasible, JAKi therapy is recommended. The type of JAKi offered is dependent on their platelet count where with platelets ≥ 50 × 109/L, options include Ruxolitinib, Fedratinib and Momelotinib. By contrast, with platelets < 50 × 109/L, Momelotinib or Pacritinib are recommended [24].
JAK Inhibitor TherapiesRuxolitinibRuxolitinib is a potent and selective oral JAK1/JAK2 inhibitor and is currently approved for higher-risk MF patients as well as lower-risk patients with high symptom burden [23]. These recommendations were based on two pivotal studies, COMFORT-I and COMFORT-II which examined Ruxolitinib in int-2 and high-risk MF patients (Table 2) [26, 27].
Table 2 Summary of JAKis in MF including clinical trialsBoth COMFORT-I and COMFORT-II demonstrated that Ruxolitinib had superior efficacy in comparison to placebo in the former and best available therapy (BAT) in the latter [26, 27]. Regarding efficacy 41.9% of patients receiving Ruxolitinib demonstrated spleen volume reduction of more than 35% (SVR35) compared to 0.7% patients in the placebo group by week 24 in COMFORT-I [26]. Similar observations were made in COMFORT-II (28% Ruxolitinib versus 0% BAT) [27]. Of interest, a recent 5-year analysis of the COMFORT-II patients demonstrated nearly all of the Ruxolitinib cohort had some degree of initial SVR, which lasted for 48 weeks or more with continued therapy, and the median response duration was 3.2 years [28].
As well as spleen reduction, Ruxolitinib reduced symptoms with > 50% reduction in MF-SAF TSS observed in 45.9% of patients at week 24 compared to 5.3% on placebo in COMFORT-I [26]. This was also noted in COMFORT-II, using the FACT-Lym and EORTC QLQ-C30 scores [27].
As the JAK2 V617F allele burden correlates with survival outcome in PMF, this was also examined in the recent 5-year analysis of the COMFORT-II study [27, 28]. At baseline 110 patients were JAK2 V617F positive with median allele burden of 84%. Over the course of treatment with Ruxolitinib, one-third of evaluable patients had a > 20% reduction, which was sustained at week 192 [28].
As patients could cross over from the control arms in both COMFORT studies, survival benefit of Ruxolitinib was not easily demonstrated. Subsequently, Vannucchi et al. [29] published a pooled analysis across both COMFORT studies which accounted for this [29]. Overall, Ruxolitinib was associated with improved median overall survival (OS) compared to either placebo or BAT (5.3 years versus 3.8 years respectively). Furthermore, all patients who achieved SVR in the Ruxolitinib group had a better prognosis compared to those with either no change or an increase in spleen size [30].
However, several Ruxolitinib-associated adverse events (AEs) were reported in both COMFORT studies, the most common being anemia and thrombocytopenia [26, 27].
Anemia is a common finding in newly diagnosed MF patients and is a negative prognostic factor [30]. Therefore, Ruxolitinib can exacerbate this further, which can hinder the initiation and maintenance of this treatment in MF patients. In COMFORT-I and COMFORT-II, grade 3/4 anemia occurred in 45.2% and 42% patients respectively, although the rate of dose modification and/or interruption was low [26, 27].
Thrombocytopenia was another common AE observed with Ruxolitinib in both COMFORT studies [26, 27]. However, contrary to anemia, the degree of thrombocytopenia did limit Ruxolitinib dosing in MF patients [26, 27, 31]. To optimize dosing and titration of Ruxolitinib in MF patients with lower platelet counts, an open-label phase 2 study examined this further in patients with platelets between 50 to 100 × 109/L. These patients were initiated on Ruxolitinib at 5 mg bid, which was titrated up to ≥ 10 mg bid by week 24 in 48% of patients. Notably, 73% of patients titrated to 10 mg bid achieved SVR35, while 44% achieved ≥ 50% reduction in MF-SAF TSS [32]. Similar findings were observed in the phase 3 JUMP study, with lowest dose 5 mg bid used in patients with platelets 50 to 100 × 109/L [33]. EXPAND was another Phase 1b dose finding study that evaluated the starting dose of Ruxolitinib in 69 MF patients with baseline platelet counts of 50–99 × 109 /L [34]. It had two strata: baseline platelet counts of 75–99 × 109/L (Stratum 1) and baseline platelets of 50–74 × 10.9/L. Maximum safe starting dose was established in both strata as 10 mg twice daily and spleen response was achieved at week 48 in 33.3% and 30.0% of patients in Stratum 1 and 2 respectively [34].
However, the observational, real-world RUXOREL-MF study highlighted the importance of dose optimization to maximize Ruxolitinib clinical benefit [35]. This examined 209 MF patients treated with Ruxolitinib and focused on disease-related and treatment-related features in the first 6 months. Risk factors for shorter survival included ≤ 30% palpable spleen reduction as well as Ruxolitinib treatment at dose < 20 mg twice daily. Based on these findings, the group developed a prognostic model, the Response to Ruxolitinib after 6 months (RR6) score, which incorporated these factors as well as red cell transfusion requirement. Unsurprisingly, high-risk patients (i.e. score ≥ 2.5) had reduced median OS of 33 months compared to low-risk patients (score 0) where median OS was not reached [35].
Non-hematological AEs have also been observed with Ruxolitinib including infections and non-melanoma skin cancers (NMSCs). Regarding the former, a meta-analysis of clinical trial data from 6 randomized controlled studies did note an increased risk of herpes zoster in MF patients [36]. In relation to NMSCs, a 10-year retrospective cohort study of 564 patients, of which 188 were exposed to Ruxolitinib, demonstrated an increased incidence of NMSCs, especially squamous cell carcinomas [37]. Rampotas et al. [38] further supported this with a UK-wide retrospective study of MF patients who developed NMSC while on Ruxolitinib. They identified 106 NMSCs in 90 patients over a > 10 year period, with an increased risk of NMSC recurrence and metastasis noted in those who continued Ruxolitinib treatment [38].
Overall, Ruxolitinib is a highly effective treatment in MF patients as demonstrated by the COMFORT-I and COMFORT-II studies. However, the development of cytopenias, especially thrombocytopenia, can limit dosing and thus influence clinical outcome in these patients. Furthermore, discontinuation rates can be high ranging from 40 to 70% during the first year of treatment, and patient outcome after Ruxolitinib discontinuation is generally poor, with median progression-free survival (PFS) of 6.0 months and OS 11.1 months [39, 40].
FedratinibFedratinib is a selective JAK2 and FMS-like tyrosine Kinase 3 (FLT3) inhibitor which was approved by the FDA for int-2 and high-risk MF patients who were Ruxolitinib naïve or resistant in 2019 [41]. These recommendations were based on results from the JAKARTA and JAKARTA-2 (Table 2). The former was a phase 3 placebo-controlled trial which evaluated once daily Fedratinib at 400 mg or 500 mg in JAKi-naïve MF patients [42]. Similar to Ruxolitinib, symptom responses at week 24 were observed in 34% of Fedratinib 400 mg patients compared to 7% placebo. Furthermore, up to 70% patients treated with Fedratinib experienced SVR at 24 weeks [42].
In JAKARTA-2, this was a phase II study which assessed the clinical activity of Fedratinib 400 mg daily in patients with int-2 or high-risk primary or secondary MF, previously treated with Ruxolitinib [43]. Notably, patients enrolled were originally classified as Ruxolitinib resistant or intolerant per investigator discretion. Furthermore, they had to have had at least 14 days of exposure to Ruxolitinib.
Initial analysis of JAKARTA-2 demonstrated increased efficacy with Fedratinib with improved spleen size and symptoms [43]. This analysis was recently updated using more stringent definitions of Ruxolitinib relapse, refractory or intolerance (i.e. Stringent Criteria Cohort). These criteria included relapse defined by Ruxolitinib treatment for ≥ 3 months with spleen regrowth, while refractory was defined by < 10% SVR from baseline at the same timepoint [44]. Overall 66 patients were included who received at least 6 cycles of Fedratinib. Responses were still maintained in this subgroup with SVR 30% and symptom response rate of 27% [44].
However, hematological AEs were reported in both JAKARTA and JAKARTA-2 including grade 3/4 anemia and thrombocytopenia [43]. Non-hematological AEs were also common, especially gastrointestinal toxicities [42, 43]. This was unsurprising given Fedratinib inhibits FLT3, and did result in dose interruption and reduction in 15% patients in JAKARTA-2 [43]. Importantly, Wernicke’s Encephalopathy (WE) was another potential AE related to Fedratinib and did result in this JAKi being placed on hold in 2013. However, a retrospective analysis did suggest that patients affected by WE had underlying predisposing conditions (e.g. malnutrition; gastrointestinal disturbance) [45]. Additionally, the prevalence of WE were lower than anticipated (0.4–0.7%). Based on these findings, the clinical hold was lifted in 2017.
Overall, Fedratinib is clinically effective and does reduce spleen size and symptom burden in patients previously treated with Ruxolitinib. However, the delivery of this drug is limited by both hematological and non-hematological AEs and requires close management. As a result, the FREEDOM and FREEDOM-2 studies were both designed with an aim to address this using prospective strategies to mitigate the gastrointestinal AEs and reduce risk of WE in int-2 and high-risk MF patients previously treated with Ruxolitinib [46, 47].
Results from the FREEDOM2 study were recently presented – this was a phase 3 randomized controlled trial with 201 patients of which 134 received Fedratinib while 67 received BAT [
Comments (0)