Study design
In this observational, real-world study, patients who received tixagevimab/cilgavimab from July 9, 2022, to December 30, 2022, in Hainan, China, were included.
The inclusion criteria were patients who received tixagevimab/cilgavimab either before the initiation of the study or were prescribed with tixagevimab/cilgavimab thereafter. The patients participating in interventional clinical trials for SARS-CoV-2 prophylactics or treatments were excluded.
The study was approved by the Ethics Committee of Ruijin-Hainan Hospital, Shanghai Jiao Tong University School of Medicine (approve no. KY2022-001). Written informed consent was obtained for the prospectively enrolled patients. The requirement for written informed consent was waived by the board for the retrospectively included patients. The study was registered with clinicaltrial.gov (NCT05917951).
Treatment and follow-up
The first dose of tixagevimab/cilgavimab was administrated before or prescribed after the initiation of the study. The date of the first administration of tixagevimab/cilgavimab was the index date, i.e., day 0 for that patient.
COVID-19 diagnosis was confirmed via reverse transcription polymerase chain reaction (RT-PCR) or rapid antigen tests, with the severity classified according to the World Health Organization (WHO) Clinical Progression Scale and the Guideline on Diagnosis and Treatment of Novel Coronavirus Pneumonia (Interim 10th Edition).
If the subject was administered tixagevimab/cilgavimab before enrolment, the data from the index date to the enrolment date was collected retrospectively. The baseline data, including demographics, baseline clinical characteristics, history of SARS-CoV-2 infection causing COVID-19, comorbidities from up to 12 months before the first administration of tixagevimab/cilgavimab, and other healthcare resource utilization was collected from the patient’s medical records. The data regarding COVID-19 diagnosis, outcomes, and adverse events (AEs) were prospectively collected for up to six months post-administration.
For prospectively enrolled patients, data collection started immediately after enrolment. The subjects were contacted remotely, by phone or text message, or returned to the site to enquire about COVID-19 risk behaviors, any positive test result for SARS-CoV-2 infection for themselves and any close contacts. If they reported a positive test result, their COVID-19 symptoms and hospitalization were further collected. The data were collected at baseline and 1, 3, and 6 months or until discontinuation, loss to follow-up, or death, whichever occurred first.
Outcomes
The primary outcomes were the proportion of individuals grouped by demographic variables of interest, individuals by clinical characteristics of interest, individuals by baseline COVID-19 vaccination status or any history of exposure to other prophylactic interventions for SARS-CoV-2 exposure prevention, individuals by baseline history of SARS-CoV-2 infection/COVID-19 diagnosis and disease severity, and individuals within priority tixagevimab/cilgavimab subpopulations of interest.
Secondary outcomes were the proportion of individuals by demographic variables of interest and clinical characteristics of interest within key basic disease subgroups, the proportion of individuals identified by baseline COVID-19 vaccination status or any history of exposure to other prophylactic interventions for SARS-CoV-2 exposure prevention within key basic disease subgroups, AEs, serious AEs (SAEs), AEs of special interest (AESIs), SARS-CoV-2 infection rates (both asymptomatic and symptomatic), the proportion of SARS-CoV-2 infection (asymptomatic or symptomatic) in close contacts, SARS-CoV-2 mortality rates, SARS-CoV-2 hospitalization rates, and SARS-CoV-2 intensive care unit (ICU) admission rates.
Statistical analysis
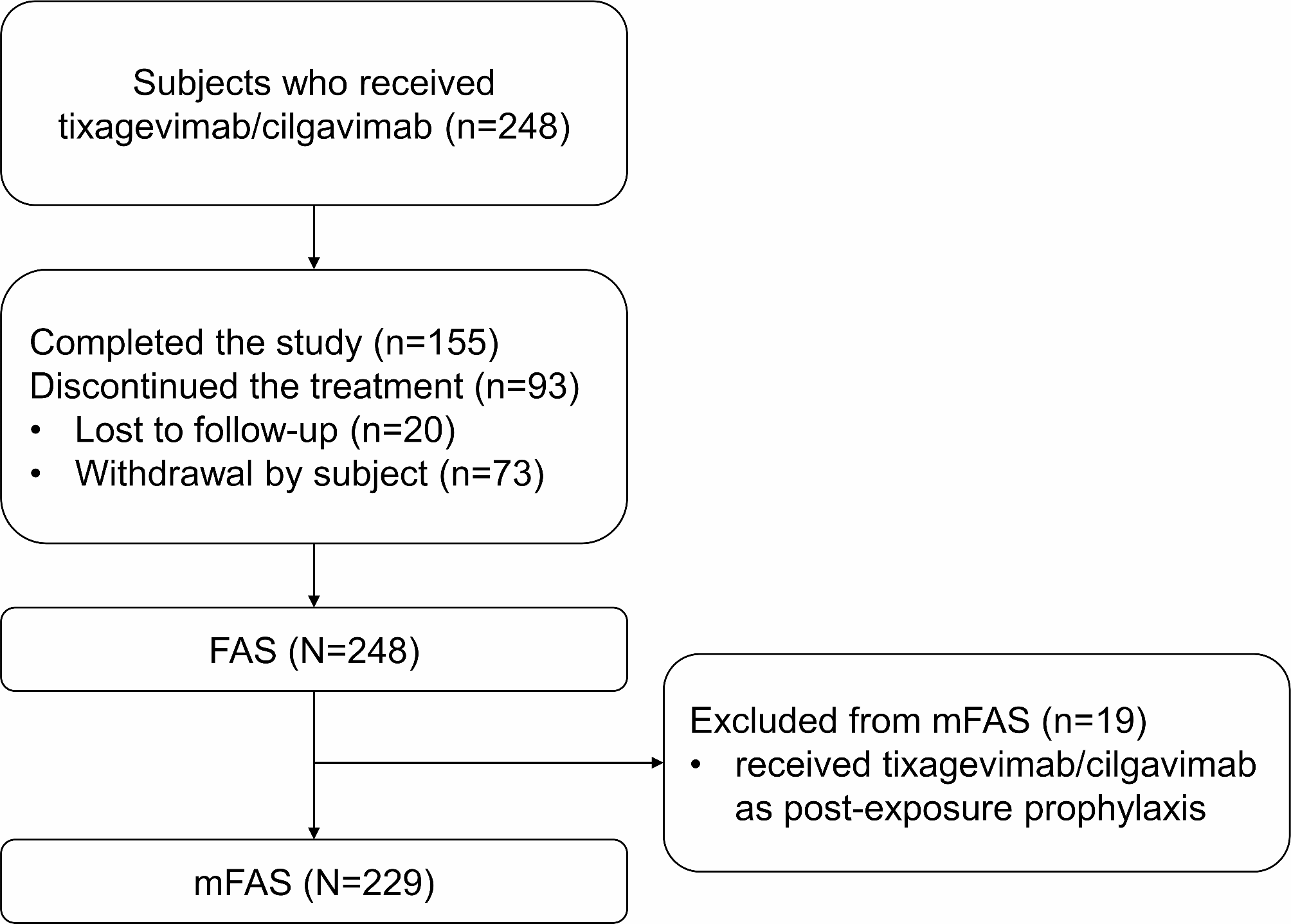
The analysis was performed using SAS 9.4 (SAS Institute, Cary, NC, USA). The full analysis set (FAS) included all patients who received at least one dose of tixagevimab/cilgavimab. The modified full analysis set (mFAS) included all patients who had received at least one dose of tixagevimab/cilgavimab for PrEP. The primary outcomes were analyzed in the mFAS.
The continuous variables were presented as mean ± standard deviation or median (range), while categorical variables were summarized as n (%). The event rates and associated exact Poisson 95% confidence intervals (CIs) were calculated. Time-to-event data were summarized using Kaplan-Meier estimates of the median event time and quartiles with their respective 95% CIs.









Comments (0)