
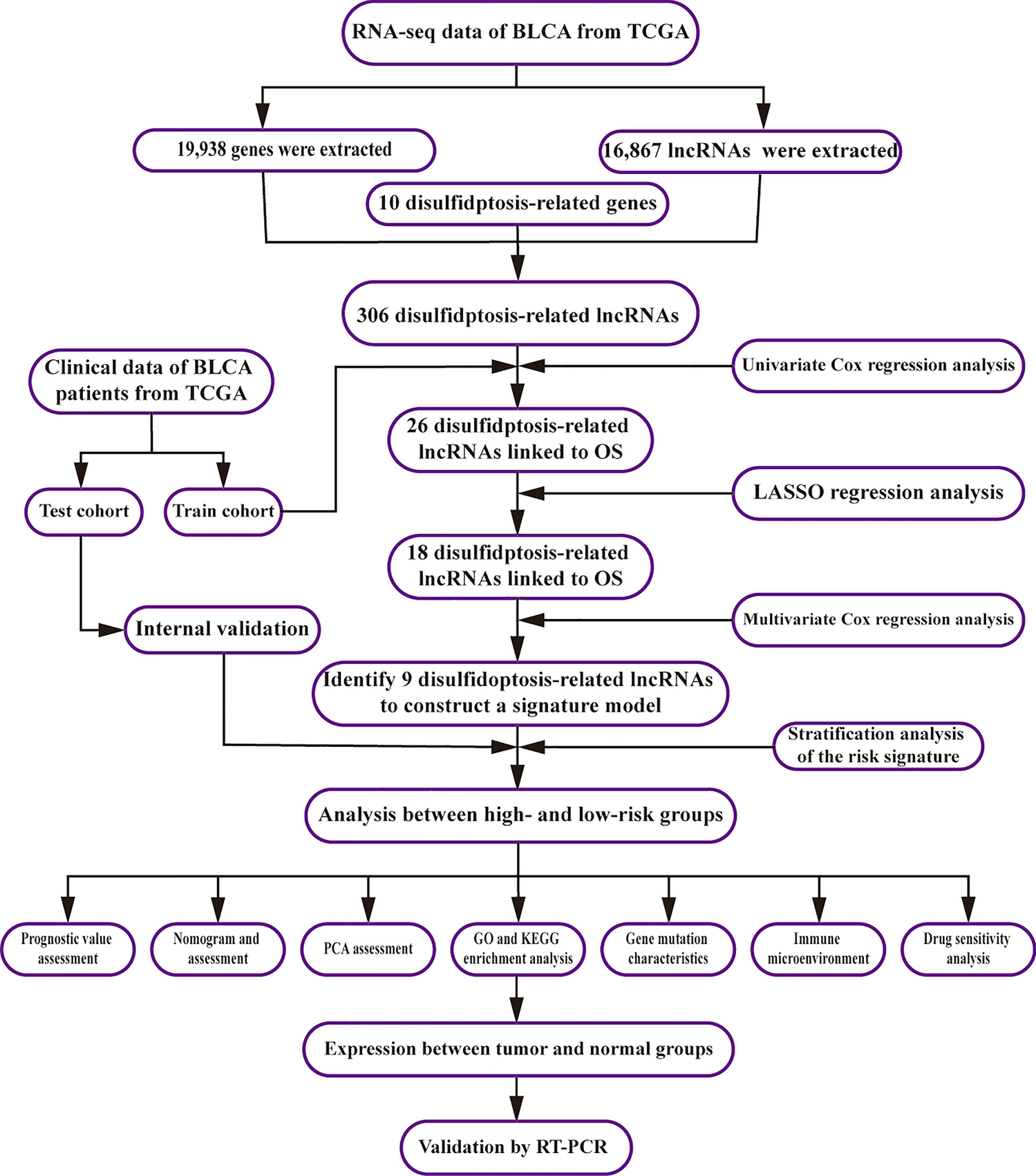
Remember me





Initially, we integrated transcriptomic data from TCGA-LGG/GBM and normal control transcriptomic data from GTEx, followed by computation using the “limma” package on TCGA-LGG/GBM. We filtered for tumor overexpressed genes meeting our criteria. Subsequently, we intersected the DEGs with Hippo pathway related genes to obtain genes overexpressed in tumors that are also associated with the Hippo pathway. Finally, we conducted univariate Cox regression analysis, through which we identified 62 genes linked to prognosis. Utilizing these 62 genes, we initially employed “ConsensusClusterPlus” for hierarchical clustering of tumor tissues. When k = 2, the clusters exhibited high cohesion and low coupling. The curve at k = 2 appeared the smoothest, aligning with our requirements, thus, we divided them into two groups, C1 and C2 (Fig. 1A). The PAC curve indicated the lowest value at k = 2 (Fig. 1B and C). Comparatively, the C2 group exhibited a poorer prognosis than the C1 group, as illustrated by the overall survival (OS) curve (P < 0.0001, Fig. 1D). To further visualize these findings, we generated a heatmap portraying the Z-scores of the 62 genes across various groups (Fig. 1E). Additionally, heatmaps representing the analysis of immune cell infiltration abundance and the expression profiles of immune modulators unveiled distinctions between the C1 and C2 groups (Fig. 1F-G).
Fig. 1
Glioma Hippo pathway-related signature (HRGs) landscapes and identification of two subclusters with distinct prognosis and immune infiltrations. A The consensus score matrix of glioma samples in TCGA-LGG/GBM when the clustering number k = 2. The consensus score represents the intensity of interaction between two samples. B,CThe CDF curves and PAC scores of the consensus matrix for each k. D The survival differences between two clusters, analyzed by Kaplan-Meier curves with the log-rank test. E The expression profiles of 62 prognostic HRGs between two clusters. F The infiltration abundances of 28 immune cell subsets evaluated by xCell, TIMER, quanTIseq, MCP-counter, and EPIC for two clusters. G The expression patterns of immunoregulators for two clusters
We conducted IC50 drug sensitivity analysis for C1 and C2 groups separately using six chemotherapeutic drugs (5-Fluorouracil, Cisplatin, Docetaxel, Gemcitabine, Gefitinib, Olaparib). We observed lower IC50 values for 5-Fluorouracil, Cisplatin, Gemcitabine, and Olaparib in the C2 group, suggesting better treatment efficacy for these drugs in the C2 group. Additionally, Docetaxel and Gefitinib were observed lower IC50 values in C1, indicating better treatment efficacy for these drugs in C1 patients (P < 0.05, Fig. 2A). Analysis of response rates to two immune checkpoint blockers (ICBs, targeting CTLA4 and PD1) between C1 and C2 groups showed higher response rates in the C1 group for ips_ctla4_neg_pd1_neg, ips_ctla4_neg_pd1_pos, ips_ctla4_pos_pd1_neg, (P < 0.05, Fig. 2B).
Fig. 2
Drug sensitivity between the two subclusters. A Violin plot displaying the estimated IC50 of chemotherapeutic agents between two clusters. B Raincloud plot displaying the IPS scores between two clusters
3.2 Weighted Gene Co-expression Network Analysis (WGCNA)We finally selected C2 subgroup with poor prognosis for WGCNA analysis. We utilized Scale Independence and Mean connectivity to screen for the soft threshold, ultimately setting it at 4 (Fig. 3A). Subsequently, we visualized the Cluster Dendrogram plot (Fig. 3B). We correlated the nine gene sets obtained from the analysis with clinical factors (futime, fustat, age, cluster). We selected several gene modules, namely MEgreen, MEblack, MEpink, MEturquoise, and MEyellow, based on their strongest correlation with the cluster (P < 0.0001, Fig. 3C). Then, employing MM = 0.6 and GS = 0.3 as criteria, we further filtered the key genes within the five gene modules (MEgreen, MEblack, MEpink, MEturquoise, MEyellow) (Fig. 3D).
Fig. 3
WGCNA identifies C2-related modules. A Analysis of network topology for different soft-threshold power. The left panel shows the impact of soft-threshold power on the scale-free topology fit index; the right panel displays the impact of soft-threshold power on the mean connectivity. B Cluster dendrogram of the coexpression modules. Each color indicates a co-expression module. C Module-trait heatmap displaying the correlation between module eigengenes and clinical traits. D Correlation between module membership and gene significance in the five modules. Dots in color were regarded as the hub genes of the corresponding module (MM > 0.6 & GS > 0.3). E Top enriched GO terms of hub genes
Finally, we carried out Gene Ontology (GO) analysis on the selected genes. According to results, among the biological process (BP) related pathways, the gene enrichment of chromosome separation pathway is the highest, with 62 genes, while the gene enrichment of chromosome separation regulation pathway is the lowest, with only 31 genes. Within Cellular Component (CC)-related pathways, the highest gene enrichment was observed in the chromosomal region, with 46 genes, whereas the outer kinetochore exhibited the fewest enriched genes, totaling 12. In Molecular Function (MF)-related pathways, the highest gene enrichment was observed in tubulin binding and microtubule binding, each with 17 genes, while collagen binding exhibited the fewest enriched genes, totaling 7 (Fig. 3E).
3.3 Generating and validating predictive signatures through the utilization of various machine learning techniquesWe employed the CGGA_693 and CGGA_325 cohorts as the validation sets, while the TCGA-LGG/GBM served as training cohort. Genes were inputted into union machine learning algorithms constructed from a combination of 10 different methods. Subsequently, we ran the machine learning algorithms on the training cohort and fitted union prediction models using a ten-fold cross-validation framework. The value of C-index for all cohorts were computed for each combination algorithm to compare the predictive capabilities of the models (Fig. 4A). Since the Stepcox[both] + LASSO algorithm shows the highest average C-index in the three sets (average C-index = 0.783), we chose it to develop our prediction model. To calculate the risk score, we first multiply the gene expression levels by their corresponding coefficients, and then sum the resulting values of all model genes. Our formula is as follows:
Fig. 4
Machine learning-based HRGs construction. A Total of 101 kinds of prediction models fitted in TCGA-LGG/GBM and verified in two validation cohorts. The model was ordered by the average of the C-index of all datasets. B Survival differences between two groups in three datasets. C Time-dependent ROC analysis of the model in three datasets
Risk score = SERPINH1 * 0.268219518 + ADAM12 * 0.093599139 + ITGA5 * ( − 0.337893272 ) + LAMB1 * ( − 166722911 ) + PLAU * (− 0.113386323 ) + COL5A2*0.114227051 + SERPINE1 * 0.048412168 + MMP11 * 0.035966659 + FAM114A1 * 0.237835006 + BGN * 0.124278238 + SEC24D * (− 0.322569217) + FN1 * 0.041024082 + ITGA4 * (− 0.040729289) + MMP14 * (− 0.017954319) + FAM20A * 0.005381167 + EMILIN1 * 0.192405744 + VDR * 0.135692503 + RDH10 *(− 0.00271331) + CLCF1 * 0.0269214 + PLAUR * 0.448904471 + NAMPT * 0.173534547 + SOCS3 * (−.025755306) BCL3* (− 0.096713155) + DPYD * 0.001112946 + FCGBP * (− 0.020501538) + S100A11 * 0.062038109 + PRSS23 * 0.007445159 + FTL * (− 0.168039335) + CASP4 * 0.340438325 + MYO1G * (− 0.267030097).
Additionally, based on the risk scores inferred by the model for each sample, we stratified each dataset into high-risk and low-risk groups according to the median for overall survival (OS) analysis. In the TCGA, CGGA_693, and CGGA_325 datasets, the high-risk group consistently had poorer outcomes than the low-risk group (P < 0.0001, Fig. 4B). In addition, from the ROC curve, we can see that the model has an AUC of over 0.7 at 1, 3, and 5 years, indicating robust diagnostic performance across all the three time intervals (Fig. 4C).
3.4 Immunological Analysis, Drug Sensitivity Analysis, and GSEA AnalysisOur immunoinfiltration analysis compared 22 groups of immune cells between two risk groups. The results of box plot exhibit the infiltration levels, indicating that the levels of immunoinfiltration of various immune-related cells such as activated B cells, activated CD4 T cells, and activated CD8 T cells were higher in the high-risk group than in the low-risk group (P < 0.01). In contrast, the low-risk group had a higher level of immunoinfiltration of eosinophilic cells (P < 0.0001, Fig. 5A). Additionally, examination of expression differences in M2 marker-related genes (CXCR2, CCL2, IL6, CXCR4, TGFB2, TGFB1, TGFB3) revealed elevated expression levels in the high-risk group (P < 0.001, Fig. 5B). Additionally, box graph exhibited that the expression levels of text-related markers (CXCL13, HAVCR2, LAG3, LAYN, PDCD1, TIGIT) were higher in high-risk group (P < 0.05, Fig. 5C).
Fig. 5
TME phenotypes between risk groups. A Box plot illustrating the distributions of 22 immune cell subsets determined by CIBERSORT between two risk groups. B, C Box plot illustrating the expression profiles of M2 polarization regulators B and TEXterm features C between two risk groups
We employed drug sensitivity analyses in two groups and included six commonly utilized chemotherapy agents (5-fluorouracil, cisplatin, docetaxel, gefitinib, gemcitabine, and olaparib). We found that the IC50 scores of 5-fluorouracil, cisplatin and gemcitabine in low-risk group were higher (P < 0.01). Conversely, the high-risk group exhibited higher IC50 scores for Docetaxel, Gefitinib, and Olaparib (P < 0.05, Fig. 6A). After conducting mRNA instability (mRNAsi) scoring on the relevant dataset, we stratified the training set into groups with high and low mRNAsi scores. The overall survival (OS) prognosis curve showed that the mRNAsi group with a higher score had a better prognosis (P < 0.0001, Fig. 6B). Examination of mRNAsi score disparities between the C1 and C2 groups unveiled higher mRNAsi scores in the C1 group (Fig. 6C). Furthermore, contrasting the low-risk and high-risk groups, we observed lower mRNAsi scores in the low-risk group (Fig. 6D).
Fig. 6
Chemotherapy sensitivity and mRNAsi between two risk groups. A Violin plot displaying the estimated IC50 of chemotherapeutic agents between two risk groups. B Survival differences between two mRNAsi groups in TCGA-LGG/GBM. C, D Violin plot displaying the mRNAsi index between two clusters (C) and two risk groups (D)
The GSEA analysis identified six upregulated cell proliferation-related pathways (Glycolysis, Angiogenesis, Epithelial Mesenchymal Transition, E2F Targets, Hypoxia, G2M Checkpoint) in the high-risk group (adjusted p-value < 0.001).We selected the top three pathways with the highest normalized enrichment score (NES) for GSEA analysis, revealing upregulation of B Cell Mediated Immunity (NES = 2.55, adj.P < 0.001), Lymphocyte Mediated Immunity (NES = 2.52, adj.P < 0.001), and Regulation of Lymphocyte Proliferation (NES = 2.40, adj.P < 0.001) in the high-risk group (Fig. 7A). Additionally, we also selected the three pathways with the lowest NES scores for GSEA analysis, and the results showed that Regulation Of Postsynaptic Membrane Potential (NES= − 2.92), Modulation Of Chemical Synaptic Transmission (NES= − 2.84), Synaptic Vesicle Cycle (NES= − 2.83) These three pathways were up-regulated in the low-expression group (adj.P < 0.001, Fig. 7B).
Fig. 7
Dysregulated cancer hallmarks A and KEGG pathways B between two risk groups
3.5 Single-cell AnalysisWe conducted dimensionality reduction clustering on three integrated single-cell datasets, GSE162631, GSE84465, and GSE138794, using the UMAP algorithm, resulting in 11 clusters (Fig. 8A). Furthermore, we annotated cells based on marker genes, identifying a total of seven cell types: Macrophage, Fibroblast, Monocyte, Astrocyte, Neutrophil, Oligodendrocyte, and B cell (Fig. 8B).
Fig. 8
The highly activated HRGs in malignant glia cells. A UMAP visualization of 78, 738 cells from three public glioma scRNA-seq cohorts. B Seven major cell types were automatically annotated by “SingleR” package. C VlnPlot illustrating the expression values of cell type-specific markers. D Malignant glia cells identification by “CopyKAT” package. The HRGs expression in single cell level determined by AddModuleScore function in “Seurat”
Violin plots displayed variations in gene expression levels among distinct cell types. Specifically, APOE exhibited the highest expression level in Neutrophils, while its expression was lowest in B cells; DCN showed the highest expression level in Fibroblasts and the lowest in Neutrophils; CD14 had the highest expression level in Monocytes and was nearly absent in Neutrophils; AQP4 displayed the highest expression level in Astrocytes and was scarcely expressed in Neutrophils; ELANE demonstrated the highest expression level in Neutrophils and the lowest in B cells; OLIG2 exhibited the highest expression level in Oligodendrocytes and the lowest in B cells; CD79A is almost not expressed in neutrophils, and the highest expression is in B cells (Fig. 8C).
Subsequently, we identified malignant cells within the glial cell population (Fig. 8D). Lastly, we evaluated our prognostic model and observed a high level of activity in malignant cells. Additionally, within the malignant cell population, there were variations in the distribution of prognostic model scores (Fig. 8E).
Comments (0)