
Remember me
All animal work was conducted according to relevant international and national guidelines for care and the humane use of animals and was approved by the LAGeSo (Landesamt für Gesundheit und Soziales) Berlin, Germany (approval number G0294-17). Animals were humanely euthanized as follows. Small chickens (up to 4 weeks of age) were stunned by a firm blow to the head and then euthanized by cervical dislocation. Larger chickens (including final necropsy) were anesthetized using a combination of ketamine (40 mg/kg) and xylazine (5 mg/kg) via intramuscular injection into the breast muscle and subsequently euthanized by cervical dislocation. All animal experiments were conducted in a blinded manner to eliminate subjectivity.
CellsChicken embryo cells (CEC) were generated from fertilized specific pathogen-free (SPF) VALO eggs (VALO BioMedia GmbH; Osterholz-Scharmbeck, Germany), following previously published methods16. CEC were cultured in Eagle’s minimal essential medium (PAN Biotech; Aidenbach, Germany), complemented with 1 to 10% fetal bovine serum (PAN Biotech) and 1% penicillin [100 U/mL]/streptomycin [100 µg/mL] (AppliChem; Darmstadt, Germany) at 37 °C and 5% CO2. The reticuloendotheliosis virus (REV)-transformed chicken T cell line 855-1917,18, was cultured in RPMI 1640 (PAN Biotech; Aidenbach, Germany) supplemented with 1% sodium pyruvate (PAN Biotech), 1% non-essential amino acids (Biochrom; Berlin, Germany), 10% fetal bovine serum and antibiotics, and maintained at 41 °C in a 5% CO2 atmosphere.
VirusesThe GaHV-3 strain SB-1 lacking its TMRs was generated using the bacterial artificial chromosome (BAC) system of SB-1 published previously19. The mini-F cassette of the BAC contains an enhanced green fluorescent protein (eGFP) and was removed for the in vivo studies. The TMRs were deleted in the SB-1 genome using two-step Red-mediated recombination as described previously20,21,22. Briefly, the TMR copies in the SB-1 genome were sequentially deleted, resulting in a virus that lacked the TMRs in the terminal repeat region (∆TMR-TR) and one that lacked both copies (∆TMR; Fig. 1A). The resulting clones were confirmed by restriction fragment length polymorphism (RFLP), Southern blotting, and Sanger sequencing. In addition, the viruses used for in vivo experiments were verified by Illumina MiSeq sequencing with more than 1000-fold coverage. This deep coverage ensures that both the parental and mutant viruses are identical, except for the intended deletion of the TMRs. Recombinant viruses were reconstituted by transfection of CEC with purified BAC DNA using calcium phosphate transfection, as described previously23. All viruses were propagated in fresh CEC (max 3–5 passages). Virus stocks were frozen in liquid nitrogen and titrated prior to their use.
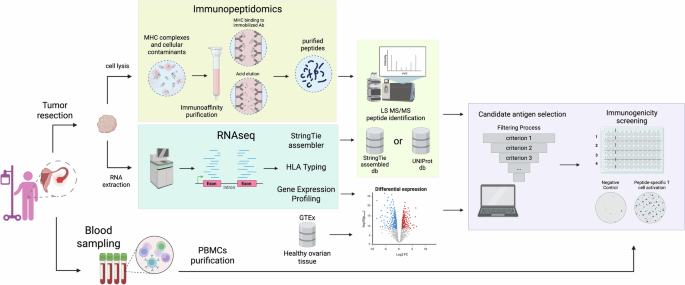
Fig. 1: Generation and characterization of the ΔTMR mutant.
A Schematic representation of the SB-1 genome with the TMR deletion in the terminal and internal repeat regions, resulting in the SB-1 ΔTMR mutant. B Restriction fragment length polymorphism patterns of the indicated viruses with the corresponding Southern blot analysis. The parental SB-1 BAC, the ΔTMRTR, and the double deletion mutant ΔTMR were digested with HindIII. The TMR sequences of the mutant viruses were detected using TMR-specific DIG-labeled probes. C Multi-step growth kinetics assays of indicated viruses. The viral genome copy numbers were quantified by qPCR. The mean copy numbers per 1 × 106 cells are presented as the averages of three independent experiments (shown as means ± standard deviations; p > 0.05, Mann–Whitney U-test). D Plaque-size assays of indicated viruses. The box plots depict the mean plaque diameters from three independent experiments, with the minimum and maximum values. Statistical analysis (p > 0.05, Student’s t-test) was performed with a sample size greater than 50.
Southern blottingTo verify the deletion of the TMRs, BAC DNA was digested with HindIII and then separated by agarose gel electrophoresis. The digested BAC DNA was transferred to a positively charged nylon membrane (Immobilon-NY+, Merck Millipore, Darmstadt, Germany) for Southern blot analysis. Fragments containing TMR arrays (Fig. 1B) were detected with a TMR-specific DIG-labeled probe (Table 1). The blots were subjected to immunological detection using an anti-DIG alkaline phosphatase-labeled antibody (Roche GmbH, Mannheim, Germany) followed by treatment with CDP-Star, ready-to-use (Roche GmbH), a chemiluminescent substrate for alkaline phosphatase.
Table 1 Primers and probes used in this studyMulti-step growth kineticsThe replication properties of the viruses were assessed by quantitative PCR (qPCR)-based multi-step growth kinetics as previously described22. Briefly, one million CEC were infected with 100 plaque-forming units (pfu) of the respective viruses. Cells were harvested at indicated time points over the course of 5 days, followed by DNA extraction using the RTP DNA/RNA Virus Mini kit (Stratec; Berlin, Germany). MDV genome copies of three independent experiments were evaluated by qPCR. Primers and probes specific to SB-1 infected cell protein 4 (ICP4) and chicken inducible nitric oxide synthase (iNOS) are listed in Table 1. Virus genome copies were normalized against the chicken iNOS gene.
Plaque-size assaysTo assess the cell-to-cell spread of the recombinant viruses, we performed plaque-size assays as described previously22. Briefly, one million CEC were infected with 100 pfu of each virus. Plaque areas were measured at 6 days post-infection (dpi) using the Bioreader (Bio-Sys; Karben, Germany), and plaque diameters were determined with the Bioreader software. Plaque-size assays were performed as three independent experiments.
In vitro integration assaysTo determine the integration efficiency of SB-1, we established an in vitro integration assay based on an assay developed for MDV24 using the chicken T cell line 855-19. One million cells were infected by co-cultivation with a highly infected CEC monolayer for 16 h as previously described25. T cells were then carefully removed, seeded into a new plate, and cultured for 14 days. The percentage of infected T cells was measured by flow cytometry using the CytoFlex S flow cytometer (Beckman Coulter, Brea, CA, USA). To assess the role of the TMRs in SB-1 genome maintenance, viral genome copies were quantified by qPCR at 1 and 14 dpi relative to cellular genome copies using specific primers and a probe for SB-1 ICP4 and the cellular iNOS gene (Table 1). Integration of SB-1 was visualized in metaphase chromosomes at 14 dpi with an SB-1-specific probe by fluorescent in situ hybridization (FISH) as described previously24,26.
In vivo characterization of the ∆TMR virusAnimal experiment 1To investigate the role of TMRs in SB-1 latency and reactivation, one-day-old SPF VALO chickens (VALO BioMedia)27 were randomly distributed into two groups and housed separately. The chickens of each group were infected subcutaneously with 2000 pfu of either the SB-1 (n = 6) or ΔTMR (n = 6). Three infected chickens per group were sacrificed at 14 and 28 dpi, respectively, and thymus, spleen, and blood samples were collected from each chicken. The lymphocytes from the spleen of each chicken were isolated using Ficoll density gradient centrifugation. Furthermore, feathers were collected from the chickens to assess the shedding of the virus. In addition, dust samples (three 1 mg aliquots) were collected from the air filters in each room at indicated time points.
Animal experiment 2To determine the role of TMRs in integration in vaccine-induced protection, one-day-old SPF VALO chickens were vaccinated subcutaneously with 2,000 pfu of SB-1 (n = 25) or ΔTMR (n = 25). Vaccinated chickens were challenged at 7 days post vaccination (dpv) by intra-abdominal inoculation of 2000 pfu of the BAC-derived very virulent plus (vv+) GaAHV2 686 strain28. Non-vaccinated chickens (n = 10) infected with GaAHV2 686 at day 7 of age were included as a control group. Whole blood samples were collected at 4, 7, 10, 14, 21, 28, and 35 dpi. The experiment was performed in a blinded manner to eliminate any subjectivity. Chickens were monitored daily for the onset of clinical symptoms. Once clinical signs were detected or at the termination of the experiment (at 91 dpv), chickens were humanely euthanized and examined for gross tumor lesions.
Virus quantification in blood samples, tissues, feather follicles, and dust samplesTo assess virus replication in vivo, DNA from whole blood samples was isolated using the NucleoSpin 96 Blood Core Kit (Macherey-Nagel; Düren, Germany) according to the manufacturer’s instructions. To evaluate the efficiency of the virus shedding, DNA was extracted through treatment of the feather pulp and dust with proteinase K at 55 °C overnight, followed by phenol:chloroform:isoamyl alcohol extraction and ethanol precipitation as described previously29,30. MDV genome copies were measured by qPCR as described above, using primers and probe sets that can differentiate between the MDV challenge virus and the SB-1 vaccine (Table 1).
Statistical analysesStatistical analyses were performed using GraphPad Prism v9 (GraphPad Software, Inc.; San Diego, CA, USA). All statistical tests can be found in the respective figure legends. Data were considered significant if p ≤ 0.05 (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Comments (0)