
Remember me




This analysis utilised data from the Generation 2 (Gen2) participants of the Raine Study, a longitudinal health study that has been following the Gen2 cohort since their prenatal stages in 1989–1991 [10, 11]. Between 2010 and 2012, when participants were approximately 20 years old, they underwent a baseline eye examination. A follow-up examination occurred in 2018–2020 when the participants were around 28 [12]. Before each eye examination, participants received a detailed explanation of the procedure and provided informed written consent. Blood samples were obtained from participants when they were aged 14 or 17. Samples were analysed in 2010 for 1,592 participants using the Infinium HD Human660W-Quad Beadchip Array, and those from an additional 310 participants were analysed in 2013 using the Infinium OmniExpress-24 BeadChip Array, for a total of 1,902 participants.
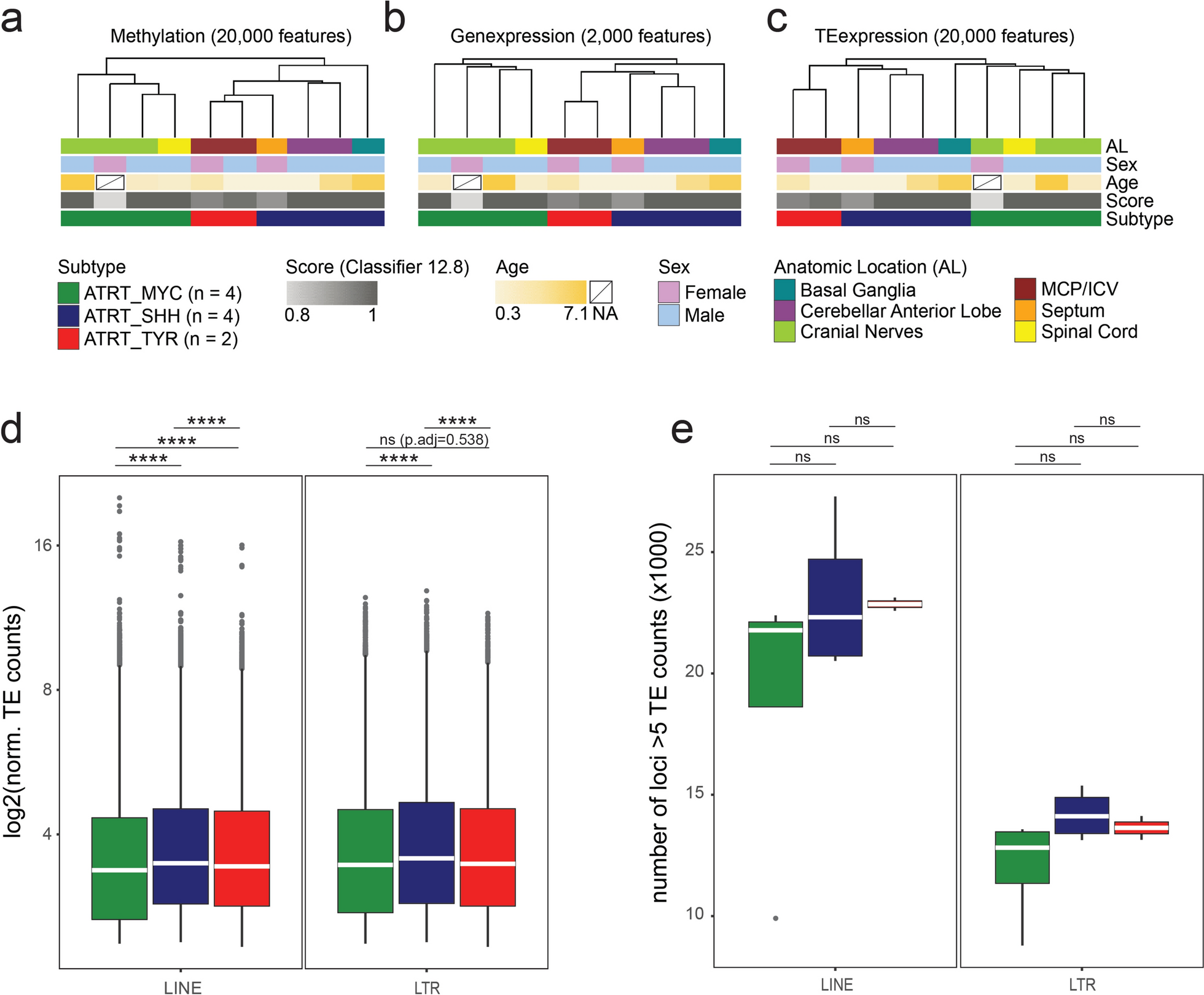
Retinal integrity estimatesParticipants underwent Spectral Domain OCT imaging (SD-OCT; Spectralis HRA + OCT, Heidelberg Engineering, Heidelberg, Germany) at the 20- and 28-year eye examinations (Fig. 1); further details outlining acquisition and processing protocols of the OCT measurements for the Raine Study are explained in Lee et al., 2020. Disc-centred 3.5-mm circular B-scans were conducted to obtain the pRNFL thickness(n = 658). A 31-slice macula-centred scan covering a 6-mm diameter area was conducted to obtain the GCIPL(n = 640) and overall macular thicknesses(n = 520) based on the Early Treatment for Diabetic Retinopathy (ETDRS) grid; the study design is outlined in Fig. 2. Outcome estimates (i.e., pRNFL, GCIPL, and overall macular thickness) were averaged between the two eyes. Cross-sectional measures for these traits at ages 20 and 28 and the longitudinal change in thicknesses between the two-time points were regressed against the PRS of PD.
Fig. 1
Spectral Domain optical coherence tomography scans centred on the disc (left) and macular (right). The 3.5 mm-diametre disc-centred B-scan obtains measurements of the peripapillary retinal nerve fibre layer thickness. The 31-slice macular-centred scans cover a 6-mm diameter area
Fig. 2
(A) Polygenic risk scoring analysis in the Raine Study (Gen2), which included two time point measurements (20 and 28 years of age) and longitudinal changes of three OCT outcome variables: GCIPL, pRNFL, and overall macular thickness. (B) Study design to evaluate the genetic overlap between ganglion-cell structural estimates (i.e., macula RNFL and GCIPL) and Parkinson’s disease
Parkinson’s disease GWAS summary statisticsWe leveraged GWAS summary statistics for a PD meta-analysis that included ~ 37.7 K cases, ~ 18.6 K UK Biobank proxy cases (having a first-degree relative with PD), and 1,417,791 controls, yielding a total sample size of 1,474,097. This dataset included samples of European ancestry from multiple cohorts, including the International Parkinson’s Disease Genomics Consortium (IPDG), 23andMe Inc., and the UK Biobank. More information about the GWAS meta-analysis is available in the corresponding publication [8]. Summary-level data from the 23andMe cohort was obtained through the corresponding application procedure (https://research.23andme.com/dataset-access/) and Institutional Data Transfer Agreement. Additionally, we obtained a version of the summary statistics excluding the 23andMe, Inc. cohort from corresponding authors [8].
Estimating polygenic risk scores for parkinson’s diseasePolygenic risk scores (PRS) is a statistical method that adds the number of risk alleles a person carries weighted by their effect sizes to estimate an individual’s genetic risk for developing a particular disease. PRS were used to evaluate the association between the genetic risk of PD and retinal integrity estimates. PRS estimates for Raine participants were derived using the GWAS summary statistics for PD described above [8] and PLINK 2.0 [13]. We selected 105 independent SNPs using the following parameters: --clump-r2 0.05, --clump-p1 5e-8, and --clump kb 1000. We employed a subset of the UK Biobank, comprising 5,000 healthy individuals, as a reference for linkage disequilibrium during the clumping process. Quality control measures excluded data with an SNP call rate below 0.95, a Hardy-Weinberg equilibrium p-value less than 10-6, and a minor allele frequency under 0.01. Post-quality control, the genotype data from the Raine Study were imputed using the Haplotype Reference Consortium reference panel. PRS was estimated in individuals of European ancestry, as determined through principal component analysis, using the 1000 Genomes Project as the reference population. To further assess the reliability of genetic scoring approaches, we calculated the PRS of GCIPL analysis based on GWAS studies [9], aiming to determine if the model was robust enough to predict GCIPL structural estimates in the Raine Study. A linear model was employed to evaluate the association between the scores generated from genome-wide significant SNPs and retinal integrity measurements. Linear models were adjusted for age, sex, principal components 1–10, and genotyping array.
Linkage disequilibrium score regression and colocalisationTo evaluate the overlap between retinal integrity estimates and PD, we used macula RNFL and GCIPL GWAS published by Currant et al. [9] based on 31,434 participants from UK Biobank. The genetic correlation between these retinal ganglion cell integrity measures and PD was evaluated using Linkage Disequilibrium Score Regression (LDSC). LDSC is a method that estimates the genetic correlation between phenotypes by analysing GWAS summary statistics while considering factors such as overlapping samples and polygenicity [14]. We used the 1000 Human Genome Project reference panel for LDSC estimations.
We subsequently contrasted the genetic makeup of GCIPL, macula RNFL, and PD by analysing data from existing literature using the GWAS pairwise approach (GWAS-PW) [15]. The GWAS-PW methodology evaluates the genetic overlap across specific genomic regions by segmenting the genome into 1,703 regions, then calculating the probability for four models: the region is exclusive to the retinal integrity estimate, it is exclusive to PD, shared by both with a shared causal variant and shared by both but without a common causal variant. For segments that suggested shared risk factors between retinal integrity estimates and PD, we used HyPrColoc. This deterministic Bayesian grouping method combines summary data to concurrently perform colocalisation among multiple traits.
Annotation and prioritisation of variantsRegions highlighted in the colocalisation analysis as regions with a shared causal variant were annotated by a gene-based association test, mBAT-combo v 1.94, a method recognised for its efficacy in identifying SNPs with masking effects [16]. Multiple testing was adjusted using the Bonferroni method, considering the total number of genes evaluated in our study (α = 0.05/6600 [genes], p < 7.57e-6).
We then leveraged omics data to explore the functional relevance of the genes that were consistent between the gene-based and the colocalisation analyses. Firstly, we utilised the summary-data-based Mendelian Randomisation (SMR) version 1.3.1 [17] to discern potential causal associations based on peripheral blood gene expression data from 2,765 participants from the Consortium for the Architecture of Gene Expression (CAGE) [18] and retinal gene expression data from 453 participants [19]. We used single-cell RNA-sequencing data from 23 retinal ganglion cell sub-groups comprising 247,520 cells [20] and over a million neurons, both exposed and not exposed to rotenone-induced oxidative stress [21]. Neurons encompass a diverse range of cells: dopaminergic neurons, serotonin transporters, astrocyte-like cells, ependymal cells, and clusters undergoing neuronal differentiation. Additionally, to account for the multiple tests, we applied the Bonferroni correction technique, taking into account the effective number of independent genes being analysed (α = 0.05/27 [genes], p < 0.001).
Lastly, we assessed the multi-omic profile of genes that were consistent between single-cell and bulk tissue gene expression using Omics Pleiotropic Association (OPERA) version 1.0.0. OPERA is a Bayesian method [22] that aims to provide further interpretation of the biological mechanisms underlying GWAS signals and prioritise molecular phenotypes. This evaluation encompasses the single-cell RNA-sequencing data, a methylation profile derived from mQTL of peripheral blood samples from 1,980 individuals [23], and eQTL information from the peripheral blood of 2,765 subjects from the CAGE Consortium [18].
Regions prioritised through the gene-based analysis were evaluated for missense variants. Missense mutations lead to single amino acid changes that can affect protein folding and are usually pathogenic. In the context of PD, missense mutations in the leucine-rich repeat kinase 2 (LRRK2) gene have been associated with familial PD [24]. However, it is unclear if they have a broader effect and play a role in the thinning of the ganglion layers of the retina. We used AlphaMissense, a machine-learning approach that uses the sequence to predict the protein structure and evaluate the pathogenicity of missense variants [25].
Tissue-specific gene expressionThe causal association between genes identified via functional annotation and PD-related alterations in visual cortex morphology were tested using the Allen Brain Atlas, which encompasses an extensive gene expression dataset specific to several brain regions [26]. Three brain models were employed, including a 55 and a 57-year-old male and a 49-year-old female. We chose these brain models due to their proximity to the age of onset for PD (40 to 65 years). Gene expression data were collected from 52 brain regions selected for their relevance to the retina and the vision system. These regions include the optic nerve, optic tract, optic chiasm, optic radiations, supraoptic decussation, oculomotor nerve, and occipital lobe in both hemispheres, as detailed in Supplementary Table 7. Fifteen brain regions with missing expression data were excluded from the analysis. The Allen Brain Atlas project facilitated gene expression quantification through fragment counts of RNA-Seq using quantitative PCR. Values were based on fluorescence or intensity measurements obtained from RNA microarrays.
Comments (0)