
Remember me

The generation of mice with loxP sites flanking exons 2 through 7 of the PNPLA2 gene has been previously described,60 and these mice were obtained from The Jackson Laboratories (Strain#024278). The Pnpla2lox/lox mice were bred with our Prx1-Cre (Prx1-CreTG/+) mice to knock down ATGL in early mesenchymal cells within the limb bud (ΔATGL). PCR analysis of genomic DNA from the ear or toe was used to confirm genotypes (primer sequence in Table S3). All mice were generated on a C57BL6/N background. During maintenance, the mice used for the conditional knockout study were kept under a standard 12-h light/dark cycle and had ad libitum access to a standard chow diet and water until 4 weeks of age. At 5 weeks of age, mice were fed a purified diet (D12450J, Research Diets) and maintained on this diet for 8 weeks. BMSCs and calvarial osteoblasts were isolated from 8- to 10-week-old C57BL6/N mice for all in vitro studies performed with or without ATGListatin. A cohort of male C57BL/6 N mice was used for metabolic cage profiling. For studies comparing light/dark cycles, male mice were sacrificed during their ‘dark’ cycle in the presence of red light. The mice were maintained in a clean environment, and all procedures were conducted in strict adherence to the Institutional Animal Care and Use Committee (IACUC) requirements at Vanderbilt University Medical Center (VUMC).
Indirect calorimetryMice were individually placed in home cages in a 12 h light/dark cycle, temperature/humidity-controlled dedicated room located in the Vanderbilt MMPC (RRID: SCIR_021939). Energy expenditure measures were obtained by indirect calorimetry (Promethion, Sable Systems, Las Vegas, NV). The calorimetry system consisted of home cages with bedding equipped with water bottles and food hoppers connected to load cells for food and water intake monitoring. All animals had ad libitum access to a purified diet and water. The air within the cages was sampled through microperforated stainless steel sampling tubes that ensure uniform cage air sampling. Promethion utilized a pull-mode negative pressure system with an excurrent flow rate set at 2 000 mL·min−1. Water vapor was continuously measured, and its dilution effect on O2 and CO2 was mathematically compensated for in the analysis stream.61 O2 consumption and CO2 production were measured for each mouse every 5 min for 30 s. Incurrent air reference values were determined every 4 cages. The respiratory exchange rate (RER) was calculated as the ratio of CO2 production to O2 consumption.
Bone marrow stromal cell isolation and osteoblast culturePrimary murine bone marrow stromal cells (BMSCs) were isolated as previously described.62 Briefly, the distal and proximal ends of the femur, tibia and iliac crests were cut open after removing adherent soft tissue. Total bone marrow, isolated by centrifugation at 12 000 r·min−1 for 30 s, was plated in complete α-MEM (α-MEM (Sigma; M0450), 10% FBS (Avantor; 89510-186), 1% penicillin/streptomycin (Sigma; P4333)). These cells were then incubated at 37 °C in the presence of 5% CO2. Iliac crests were not used for isolating BMSCs from the control and ΔATGL strains. As per the plastic adherence theory, the nonadherent hematopoietic cells were washed away, whereas the adherent mesenchymal stromal cells adhered to the plastic flask.63,64 This adherent population was trypsinized after 48 h, counted and plated in appropriate tissue culture-treated plates at appropriate numbers. BMSCs were then cultured in osteogenic medium (complete α-MEM, 50 μg·mL−1 ascorbic acid (Sigma; A4544), and 5 mmol·L−1 β-glycerol phosphate (Sigma; G9422)) to induce osteoblast differentiation after the cells became 80% confluent.62 For the experiment performed in the absence of osteogenic medium, cells were kept in culture in complete α-MEM medium without ascorbic acid and β-glycerol phosphate.
Calvarial osteoblast isolation and cultureCalvarial osteoblasts were isolated as previously described.65 Briefly, 5-day-old pups were euthanized by quick decapitation, and the calvaria were isolated, cleaned and transferred to sterile PBS. They were then digested in 10 mg·mL−1 Collagenase A solution 5 times, keeping them in a 37 °C shaker for 20 minutes for the first two digestions and 30 minutes for the last three. The last three digests were collected, and the cells were cultured in complete α-MEM medium. These cells were trypsinized after 48 h, counted and plated in appropriate tissue culture-treated plates at appropriate numbers and cultured in osteogenic medium.
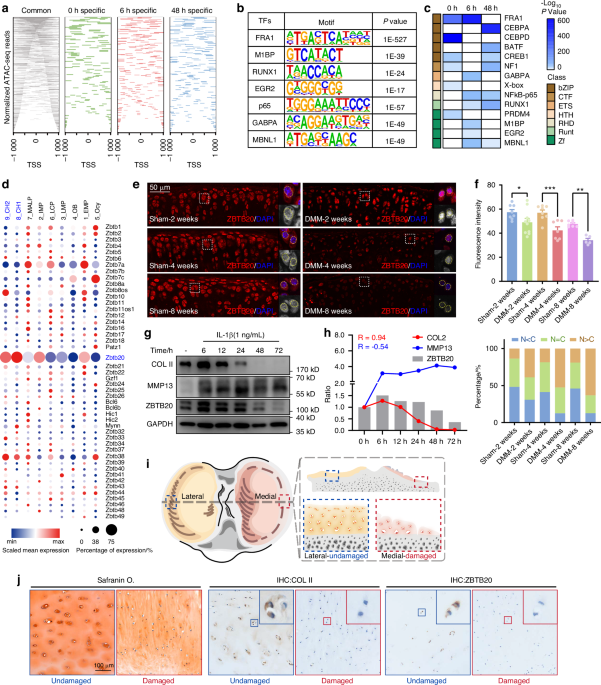
Immunostaining, imaging, and analysisBMSCs were seeded on collagen-treated glass coverslips at a density of 1.75× 105 cells per mL and grown in the presence of osteogenic medium. The cells were fixed at the mentioned time points using neutral-buffered, methanol-free 4% formaldehyde for 20 minutes. For specific experiments, cells were cultured in the presence or absence of 200 μmol·L−1 oleic acid (Sigma; O3008) in cell culture medium for 48 h. To investigate the effect of ATGListatin or etomoxir, cells were incubated with 50 μmol·L−1 ATGListatin (Signa; SML1075), 10 μmol·L−1 etomoxir (Sigma; E1905) or DMSO (vehicle) for 6 h or 3 h, respectively, before fixing and staining at the indicated time points during differentiation. The concentration of ATGListatin used was 50 μmol·L−1 for all experiments unless mentioned otherwise. The cells were then washed thrice in 1× phosphate-buffered saline (PBS) and then stained for lipid droplets using 10 μmol·L−1 BODIPY 493/503 (Thermo Fisher; D-3922) solution for 1 h at room temperature before being washed three more times with 1× PBS. For immunostaining with Runx2, after fixation, the cells were blocked and permeabilized in 0.2% gelatin B (Sigma; G-9391) with 0.1% saponin (Sigma; 47036) in PBS for 10 minutes. Immunostaining with Runx2 (Cell Signaling Technology; D1L7F) was performed overnight in 0.2% gelatin B with 0.01% saponin at 4 °C. After washing three times with PBS, the cells were stained with secondary antibody (Invitrogen; Alexa Fluor goat anti-rabbit 647; A32733) in the same buffer for 1 h at room temperature. Finally, the cells were washed thrice in PBS and once in distilled water and then mounted on slides. The coverslips were mounted on slides using Prolong Glass Antifade Mountant with NucBlue (Thermo Fisher; P36981). Confocal Z stacks with 0.30 mm thickness were taken using a Zeiss LSM 880. Lipid droplets were identified and counted using a built-in object identification program in Gen5 software. The same program was used to measure the intensity and size of these identified lipid droplets.
RNA isolationFor BMSC and osteoblast experiments, cells were seeded at a density of 5.0 ×105 per mL, cultured in osteogenic medium in the absence or presence of ATGListatin in some instances, and harvested at the indicated time points during differentiation for total RNA. RNA was isolated from cells either by a kit-based method using the ReliaPrep RNA Cell Miniprep system (Promega; Z6010) following the manufacturer’s protocol for RNA sequencing or by an organic precipitation method following lysis of the cells with TRIzol (Ambion; 15596018) for validation by quantitative real-time PCR. For the RNA sequencing experiment, BMSCs were isolated from male mice. For bone RNA extraction, flash frozen, flushed femur cortex (devoid of marrow elements) was pulverized using a Freezer Mill. Total RNA was isolated using the organic precipitation method following lysis with TRIzol. The precipitated RNA was further cleaned up using a clean-up kit from Qiagen (RNeasy MinElute Cleanup Kit; 74204). For adipose tissue RNA extraction, adipose tissue was lysed in TRIzol using a handheld homogenizer (Fisher; Homogenizer 850). RNA was isolated from the lysed tissue by organic precipitation. Isolated RNA was qualitatively and quantitatively estimated using Nanodrop One (Thermo Fisher) before making downstream applications.
Next-gen sequencingThe Vanderbilt Technologies for Advanced Genomics (VANTAGE) Core performed QC analysis of the RNA using the Agilent Bioanalyzer and an RNA Qubit assay. RNA-Seq libraries were prepared using 200 ng of total RNA and the NEBNext rRNA Depletion Kit (NEB, Cat: E6310X) per the manufacturer’s instructions. This kit employs an RNaseH-based method to deplete both cytoplasmic (5 S rRNA, 5.8 S rRNA, 18 S rRNA and 28 S rRNA) and mitochondrial ribosomal RNA (12 S rRNA and 16 S rRNA) from human, mouse, and rat total RNA preparations. The mRNA was enriched via poly-A-selection using oligoDT beads, and then the RNA was thermally fragmented and converted to cDNA. The cDNA was adenylated for adaptor ligation and PCR amplification. Sequencing was conducted using an Illumina HiSeq 2500. All samples were indexed, and multiplex sequencing was conducted on the HiSeq to generate the datasets with the customer-specified sequence yield and read length in either single or paired end format. The raw data were submitted to VANGARD for analysis. Adapters were trimmed by Cutadapt (v2.10). After trimming, reads were mapped to the mouse genome GRCm38.p6 using STAR (v2.7.8a) and quantified by feature counts (v2.0.2). DESeq2 (v.1.30.1) was used to assess differential expression between two groups. WebGestaltR (v0.4.4) was used to perform functional enrichment analysis against the Gene Ontology and KEGG databases. GSEA (v4.2.3) was used to find enriched pathways against hallmark gene sets in MSigDB (v7.5.1).
qRT‒PCR analysisComplementary DNA (cDNA; 12.5 ng) was prepared from purified RNA (Thermo Fisher; 4374967) and utilized for downstream applications. Expression analysis of selected genes by quantitative real-time PCR (qRT‒PCR) with SYBR green (Thermo Fisher; PowerUP SYBR Green; A25742) on a QuantStudio 5 (Thermo Fisher) with the following conditions: 2 min at 50 °C, 10 min at 95 °C, (15 s at 95 °C, 1 min at 60 °C) × 40 cycles followed by dissociation curve analysis (15 s at 95 °C, 1 min at 60 °C, 15 s at 95 °C). Analysis was performed by normalizing the expression of the target gene to the housekeeping gene Hprt for BMSCs and nonbone tissues and B2m for bone expression within the same sample to determine ΔCt. The ΔCt was transformed (2−ΔCt). For determining normalized to control/DMSO expression of target genes (2−ΔCt) with treatment/average of (2−ΔCt) in control/DMSO group” was determined. The sequences of primers used are listed in Table S3. To validate primer efficiencies, a concentration gradient of cDNA was used as a template.
Fluorescent fatty acid pulse chase for measuring cellular lipolysisCells seeded at a density of 5×105 per mL were incubated with complete α-MEM containing 2 μmol·L−1 BODIPY 558/568 C12 (Thermo Fisher; D3835) for 16 h at the indicated time points during osteoblastogenesis. The cells were then washed three times with 1× PBS and chased for 6 h in α-MEM medium with 2% fatty acid-free BSA (Sigma; A8806) and 1% penicillin/streptomycin 10 μmol·L−1 Triacsin C (fatty acyl CoA synthetase inhibitor, Sigma; T4540) in the presence or absence of ATGListatin. The cells were washed twice in 1× PBS and lysed in 1% Triton X-100 for lipid extraction. Lipids were extracted following the Bligh and Dyer method.66 Briefly, four volumes of chloroform:methanol (1:2) (Sigma; 319988, 179337) were added to the lysate, and the mixture was vortexed. One volume of 50 mmol·L−1 citric acid (Sigma; 251275), one volume of distilled water and one volume of chloroform were added sequentially and again vortexed. This final mixture was then centrifuged at 10 000 r·min−1 for 10 minutes. The lower organic phase was isolated and dried. The dried lipid extract was resuspended in a chloroform:methanol (2:1) mixture and loaded on TLC (Sigma; 1.05553.0001). Lipids were separated by developing TLC at room temperature in a solvent system of cyclohexane:ethyl acetate (1:2)67 (Sigma; 34855,650528) along with a triglyceride standard (Avanti Polar Lipids; 810272 P), and fluorescent lipids were visualized using iBright 1500 (Thermo Fisher). Fiji was used to analyze the fluorescent lipid bands, and the integrated density of the lipid bands from each sample was normalized to the integrated density for the origin of that sample.
Osteoblast von Kossa stainingOsteoblasts differentiated in the presence or absence of ATGListatin during the indicated time frame were assessed for mineralization by Von Kossa (VK) staining 10 days after ex vivo differentiation. Briefly, cells were washed three times in 1× PBS and then fixed with 10% neutral-buffered formalin (NBF) for 15 minutes at 37 °C. The fixed cells were then incubated in 5% silver nitrate in the presence of UV for 1 h. After 1 h, the cells were washed with distilled water and incubated with 5% sodium thiosulfate for 3 minutes. The cells were washed three times in distilled water, dried and imaged.
Fluorescent fatty acid pulse chase for investigating mitochondrial localizationBMSCs were seeded on collagen-treated glass coverslips at a density of 1.75× 105 per mL and grown in the presence of osteogenic medium. The cells were incubated with complete α-MEM containing 2 μmol·L−1 BODIPY 558/568 C12 (Thermo Fisher; D3835) for 16 h at the indicated time points during osteoblastogenesis. The cells were then washed three times with 1× PBS and chased for 6 h in complete medium with or without ATGListatin. After 6 h, the cells were washed and stained with 100 nmol·L−1 MitoTracker Deep Red FM (M22426) in incomplete medium for 30 minutes, washed with PBS, fixed in neutral-buffered, methanol-free 4% formaldehyde for 20 minutes and finally mounted on slides with Prolong Glass Antifade Mountant with NucBlue (Thermo Fisher; P36981). Confocal Z stacks with 0.30 mm thickness were taken using a Zeiss LSM 880. Colocalization analysis was performed on a per cell basis from a single stack with maximum mitochondria using ImageJ to calculate Mander’s coefficient using the threshold (tM2), where channel 1 was far red (644/665) for Mito Tracker Deep Red, denoting mitochondria, and channel 2 was red (558/568) for C12 fatty acids.
ATP rate assayTo measure ATP produced by either glycolysis or oxidative phosphorylation in real time with a Seahorse ATP rate assay, BMSCs were plated in Seahorse XFe 96-well plates at 2.5 × 104 cells per well and cultured under osteogenic conditions. At specified timepoints, ATP rate assays were performed (Agilent; 103592-100). Briefly, after removing the cell culture medium, the cells were washed with either basal assay DMEM (Agilent; 103575-100), and then the assay was performed in the absence or presence of 1, 5 or 10 mmol·L−1 glucose (Agilent; 103577-100), 2 mmol·L−1 glutamine (Agilent; 103579-100), 1 mmol·L−1 sodium pyruvate (103578-100), 200 nmol·L−1 insulin (Sigma; I9278), 60 μmol·L−1 oleic acid-BSA (Sigma; O3008), or 0.5 mmol·L−1 carnitine or in the presence of only 100 μmol·L−1 oleic acid in that same medium. For specific experiments, cells were cultured in the presence of 200 μmol·L−1 oleic acid in cell culture medium for 48 h. Subsequently, a final concentration of 2 μmol·L−1 oligomycin (Sigma; 75351) and 1 μmol·L−1 rotenone/1 μmol·L−1 antimycin A (Sigma; R8875, A8674) were injected during assays through port A and port B, while oxygen consumption rates (OCRs) and extracellular acidification rates (ECARs) were monitored in real time. For specific experiments, cells were incubated with 10 μmol·L−1 etomoxir or 5 μmol·L−1 UK5099 (Sigma; PZ0160) for 1 h in the assay medium before starting the assay or with or without ATGListatin for 24 h in cell culture medium or with both ATGListatin for 24 h in cell culture medium and etomoxir for 1 h in assay medium before the assay. Under both conditions, the assays were performed in the presence of the inhibitors in the assay medium. Considering the stoichiometry of the glycolytic pathway, the percentage of glycolysis was based mainly on the ECAR. Conversely, the rate of oxygen consumption that is coupled to ATP production during oxidative phosphorylation was calculated as the OCR that was inhibited by addition of the ATP synthase inhibitor oligomycin. This assay measured the flux of both H+ production, as indicated in ECAR, and O2 consumption, reported as OCR, simultaneously. By obtaining these data under basal conditions and after serial addition of mitochondrial inhibitors (oligomycin and rotenone/antimycin A), the rate of glycolysis and oxidative phosphorylation were able to be measured. Hoechst 33342 stain (Thermo Scientific; 62249) was also injected in the last port, and a Cytation 5 (BioTek) was used to provide cell counts, both for normalization and to monitor proliferation throughout differentiation.
Dual-energy X-ray absorptiometryDual-energy X-ray absorptiometry (DXA) was performed on the control and ΔATGL mice 8 weeks after starting the purified diet before their sacrifice in the prone position by using a Faxitron UltraFocus (Hologic). The instrument was calibrated for offset images, dark images, and flat-field images before the measurement by a method provided by the manufacturer.
Micro-computed tomography (μCT)Tibiae from the mice were cleaned of soft adhering tissue, placed in 10% neutral-buffered formalin (NBF) for 48 h, and then stored in 70% ethanol. The proximal and mid-diaphyses of these tibiae were imaged using an ex vivo micro-computed tomography (μCT) scanner (μCT 50, Scanco Medical AG, Brüttisellen, Switzerland). With a peak X-ray tube intensity and current of 70 kVp and 114 mA, respectively, 500 projections per full rotation of the sample, and an integration time of 300 ms, image stacks with an isotropic voxel size of 6 μm were acquired for the tibia metaphysis and diaphysis (310 slices each). Trabecular bone was analyzed by identifying a region of interest (ROI) 180 μm distal from the proximal tibia growth plate to include a region of secondary spongiosa extending distally 1.2 mm. Cortical bone was analyzed to include 1.2 mm ending at the tibiofibular junction.
Lipid isolation from bone and TLC analysisThe flushed tibia cortex (devoid of marrow elements) was pulverized and used to harvest total lipid by the Bligh and Dyer method66 mentioned previously. However, the powder obtained from the total flushed tibia after pulverization was weighed before adding a chloroform:methanol (1:2) mixture for normalization. Additionally, the powder was kept overnight in chloroform:methanol mixtures in a 37 °C water bath for better and complete extraction before the following steps were performed. Finally, the dried lipid extract was resuspended in an equal volume of (80 μL) chloroform: methanol (2:1) mixture and loaded on TLC (Sigma; 1.05553.0001). Lipids were separated by developing TLC at 4 °C in a solvent system of hexanes:diethyl ether:acetic acid (70:30:1) (Sigma; 293253, Emparta; 1.07026.2500, Sigma; 695092). The TLCs were stained using 10% (wt/vol) copper sulfate in an 8% (vol/vol) phosphoric acid solution and then charred at 120 °C for visualization of lipids. Fiji was used to analyze the lipid bands, and the integrated density of the lipid bands from each sample was normalized to the weight of the powdered bone sample.
Protein isolation and Western blot analysisFor BMSC and osteoblast experiments, cells were seeded at a density of 5.0 ×105 per mL, cultured in osteogenic medium in the absence or presence of ATGListatin in some instances, and harvested at the indicated time points during differentiation for total protein. Protein was isolated from cells by lysing the cells with 1× RIPA (Cell Signaling Technology; 9806) buffer in the presence of protease inhibitor cocktail (Roche; 04693116001) and phosphatase inhibitor cocktail (Roche; 04906845001). For bone protein extraction, flash-frozen, flushed femur cortex (devoid of marrow elements) was pulverized using a Freezer Mill. Total RNA was isolated following lysis with 1× RIPA buffer in the presence of protease inhibitor and phosphatase inhibitor cocktail. Protein estimation was performed using the BCA method. Equal amounts of protein (15-35 µg) from each sample were loaded and resolved using sodium dodecyl sulfate‒polyacrylamide gel electrophoresis (SDS‒PAGE), followed by transfer to PVDF membranes (Bio-Rad; 1704156). The membrane was incubated with no-stain protein labeling reagent (Invitrogen; A44449) for normalization and assessment of equal loading and finally probed for specific proteins ATGL (Cell Signaling Technology; 2439), AMPKα (Cell Signaling Technology; 5831) or phospho-AMPKα (Thr172; Cell Signaling Technology; 2535) antibodies.
HistologyThe eWAT (epididymal white adipose tissue) collected from the control and knockout mice was fixed in 10% neutral-buffered formalin (NBF) for 48 h and then stored in 70% ethanol. These samples were submitted to the Translational Pathology Shared Resource (TPSR) core for further processing. Briefly, these samples were subjected to sequential ethanol dehydration followed by embedding in paraffin. The embedded tissues were then sectioned on glass slides at 5 µm thickness, deparaffinized, and subjected to H & E staining. The number and volume of the adipocytes present in the tissue section was further quantified post microscopy imaging using BioQuant® Osteo 2018 version 18.2.6 (BioQuant® Image Analysis Corporation, Nashville, TN).
Bone histomorphometryDynamic bone formation was assessed by prior injection of sequential doses of calcein (10 mg·kg−1 body weight) and alizarin (1,2-dihydroxyanthraquinone) (30 mg·kg−1 body weight) 7 and 2 days prior to sacrifice, respectively (5-day interval). and embedded in methylmethacrylate (MMA). Briefly, the cleaned tibias were subjected to sequential acetone dehydration, and the dehydrated tibias were infiltrated using a mixture of destabilized methlymethacrylate (90%), dibutylphthalate (10%) and benzoyl peroxide (0.05%) for 3 days at 4 °C. The infiltrated bones were embedded in embedding solution, which was a mixture of the same chemicals with different concentrations of the abovementioned chemicals (85%, 15% and 4%) for 3-4 days in a 37 °C incubator. The embedding was done by pouring the embedding solution on top of base made up of 5% benzoyl peroxide in infiltration solution before the bones were placed in it. Then, the bones were embedded in MMA plastic, sectioned (5 µm) on a transverse plane and subjected to downstream analysis. Single or double labels were measured to quantify MS/BS and BFR/BS. The bone sections were stained with trichrome stain. The number of osteoblasts and osteoclasts per bone perimeter were measured at standardized sites under the growth plate at a magnification of 20 ×. Osteoblasts were identified as plump cuboidal cells lining the trabecular bone surface ( > 3 tough cells), osteoclasts were identified as large, multinucleated cells on the bone surface (mostly near the eroded surface), and bone marrow adipocytes were identified as white ‘ghost cells’ within the marrow area excluding vasculature and/or sectioning artifacts. Analyses were performed using BioQuant® Osteo 2018 version 18.2.6 (BioQuant® Image Analysis Corporation, Nashville, TN). All parameters and regions of analysis are reported as per the guidelines of the nomenclature committee of the American Society of Bone and Mineral Research (ASBMR).68,69
Serum lipid analysesBlood was collected from the carotid arteries of mice immediately before termination. Serum was isolated by allowing blood to clot for 30 min at room temperature, followed by centrifugation at 4 °C at 3 000 r·min−1 for 10 min. Serum was submitted to Vanderbilt Analytical Service Core (VASC) for further lipid analysis. Briefly, triglycerides and cholesterol were measured by standard enzymatic assays. FFAs were measured with a commercially available enzymatic kit from Fujifilm Healthcare Solutions (HR Series NEFA-HR).
Fasting blood glucoseMice were fasted for 6 h prior to testing. Following this period, blood was collected via a tail nick, and blood glucose was determined using an AlphaTRAK2 glucometer.
Transmission electron microscopyFreshly isolated tibiae were fixed in 2.5% glutaraldehyde/2% paraformaldehyde for 48 h and then decalcified (Thermo Scientific; 8340-1). The tibiae were then washed 3 times for 5 min each in 0.1 mol·L−1 cacodylate buffer before being subjected to sequential postfixation in 1% tannic acid and then 1% osmium tetroxide and en bloc staining with 1% uranyl acetate for 60 min each. The samples were dehydrated by passing through a graded ethanol series. Tissues were infiltrated with Epon-812 using propylene oxide as the transition solvent and polymerized at 60 °C for 48 h with resin for five days. Thin sections (70 nm) were cut using a Leica UC7 and a Diatome diamond knife. Thin sections were positioned on grids and stained with uranyl acetate and Reynold’s lead citrate. These sections were then imaged on a Tecani T12 TEM operating at 100 keV using an AMT CMOS camera.
Statistical analysisStatistical analyses were performed in GraphPad Prism V9. The normal distribution assumption for sample size < 40 was evaluated using the Shapiro‒Wilk normality test. Statistically significant differences across two groups were evaluated using Student’s two-tailed unpaired t test with a significance defined as P < 0.05 when normal distribution was met; when normality assumptions were not met, an alternative nonparametric test (Mann‒Whitney test) was used. The data are expressed are either the mean ± standard deviation (SD) or the mean ± standard error of mean (SEM) as described. Micro-CT analysis and RNA sequencing were performed with 5 mice from each group. For cell culture experiments, stromal cells were obtained and pooled from 6 mice in each group. For the statistical analysis of RNA sequencing, see the “RNA isolation” and “Next-gen sequencing” sections above.
Comments (0)