
Remember me
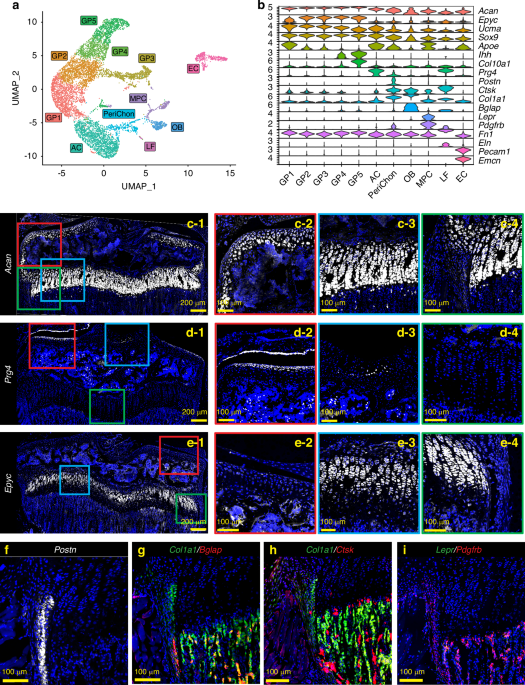
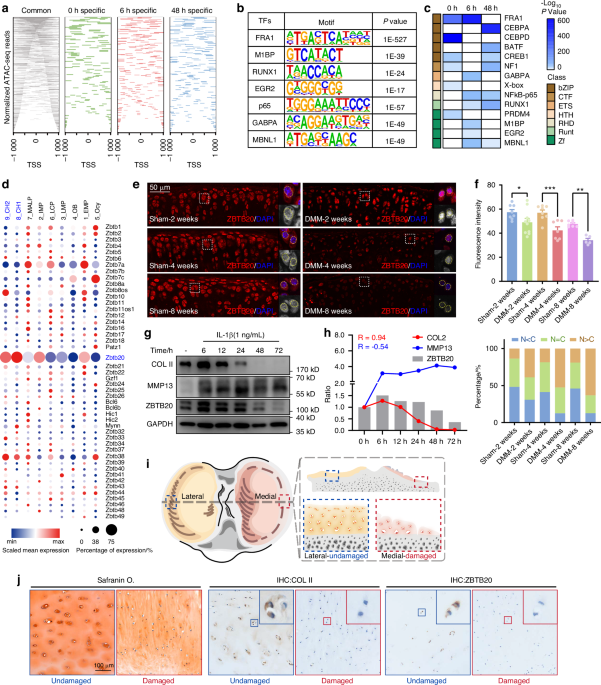

Although recent advances in scRNAseq have allowed transcriptional interrogation of cells within the bone marrow, technical and analytical approaches have a limited ability to place these cellular changes within the context of their native bone environment. To overcome this limitation, we subjected adult mouse femurs to spatial transcriptomic analyses (Fig. 1). Femurs were bisected and fixed in buffered formalin overnight at 4 °C. The samples were then decalcified in EDTA for 2 weeks with fresh decal added every 2 days. The decalcified femurs were then processed for standard paraffin embedding. Histological sections were placed on the Visium Spatial Gene Expression slide, which contains spatially unique capture oligos. Following staining and imaging, histological sections were hybridized using a comprehensive mouse whole transcriptome probe set mapping to ~20 000 genes. Following probe ligation and rinsing, specifically hybridized probes were then released from the tissue through permeabilization and captured by Visium slide oligos. Subsequent sequencing, alignment, and registration to the H&E-stained image resulted in gene expression information within its original 2D position within the bone. Following exclusion of surrounding soft tissue, as well as cartilage from the growth plate, quality controls were applied to assess the transcript recovery efficiency (Fig. 1a). To determine tissue-specific gene changes, we manually segmented spatial samples into cortical bone (orange), trabecular bone (blue), and bone marrow (magenta) (Fig. 1b). Combined, our assessment showed an average of 172 ± 154 (cortical), 194 ± 96 (trabecular), and 233 ± 131 (marrow) unique genes per spot for a combined 13 115 (cortical), 9 605 (trabecular), and 17 524 (marrow) total unique genes represented within each of the morphologically unique regions. Notably, spatial spots were assigned to the tissue most represented within each spot and, as a result, may partially contain cells from neighboring tissue regions (i.e., spots designated as “cortical bone” overlay the cortical bone by >50% but may also partially overlap with neighboring bone marrow). Gene expression profiles were used to confirm the successful spatial delineation of captured transcripts (Fig. 1c). Both cortical and trabecular bone showed high levels of mature osteogenic transcripts (Col1a1, Bglap), with additional periosteal (Postn) and osteoprogenitor (Sp7, Runx2) genes enriched within the cortical and trabecular bone, respectively. In contrast, hematopoietic (Ptprc, encoding CD45), erythropoietic (Hba-a2), and proliferative markers were heavily enriched within the bone marrow (Fig. 1c). These data show for the first time the technical feasibility of conducting spatial transcriptomics within fully mineralized adult long bone tissue.
Fig. 1
Spatial transcriptomic analyses of adult femurs. a Spatial feature plots of H&E-stained sections overlaid with the number of unique genes (nFeature) or unique transcripts (nCount) per spatial spot. b Manual segmentation of cortical bone, trabecular bone, and bone marrow spatial spots by histological morphology. c Marker genes showing enriched expression within each segmented compartment
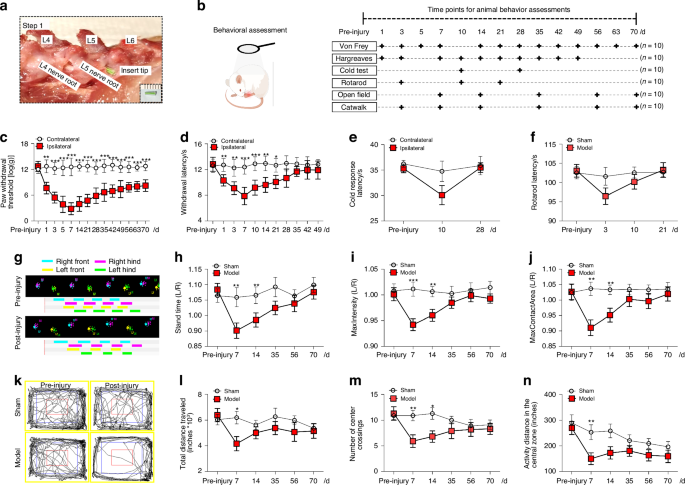
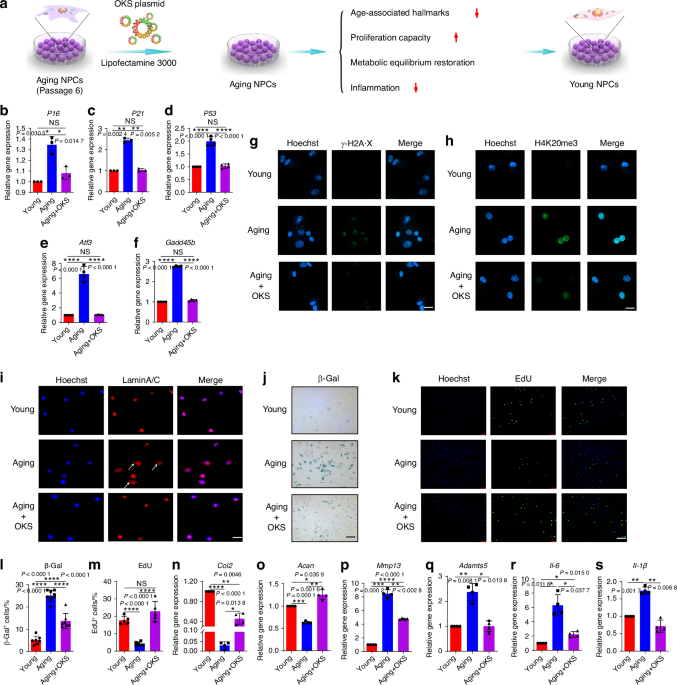
Computational deconvolution of spatial spots using scRNAseqThe high complexity of the bone marrow in terms of the diversity of cell types present in close proximity of each other, in combination with the limited spatial resolution of spatial transcriptomics, make it challenging to assign spatial gene expression profiles to individual cell types. To overcome this, we made use of scRNAseq to computationally deconvolve spatial spots into their cellular constituents. Deconvolution of spatial spots was conducted using scRNAseq datasets and the packages Seurat, CellTrek, and Cell2Location (Fig. 2). Seurat relies on data transfer of uniquely expressed genes between cellular clusters, CellTrek uses a mutual nearest neighbor-based approach to map individual cells to spatial data, and Cell2Location uses a decomposition-based approach. For our studies, multiple scRNAseq datasets were combined from bone- and marrow-derived cells5,7 to generate a single object comprising all major cell types of the bone marrow (Figs. 3a and S1). Seurat, CellTrek, and Cell2Location each provided a probability in which an scRNAseq cluster of interest is present within each spatial spot (Fig. 3b). Predictive modeling was conducted for all clusters and mapped to all regions of our spatial data. The discerning power of each predictive model was then determined by calculating the standard deviation in predictive scores within a single spatial spot across all clusters (larger values denote an increased ability to distinguish the enrichment of an individual cell type above random mixing) (Fig. 3c). Next, predictive values were interrogated in terms of their cellular placement within the different spatial compartments (Fig. 3d). These results suggest that each of the predictive models possesses positive and negative attributes. Seurat showed a relatively high abundance of mesenchymal lineage (MesLin) cells within the cortical (65%) and trabecular (44%) bone, as well as within the marrow (49%). Conversely, Cell2Location showed a high abundance of hematopoietic cells within the marrow (63%) but a disproportional abundance of smooth muscle cells (SMCs) within both cortical (44% vs. 11% and 4% in Seurat and CellTrek, respectively) and trabecular (33% vs. 4% and 21% in Seurat and CellTrek, respectively) bone regions. CellTrek appeared to perform more moderately in both assigning MesLin cells to bone compartments and hematopoietic cells to the marrow compared to Seurat and Cell2Location (Fig. 3d). To overcome these limitations within each predictive package, we created a combined predictive pipeline. All spatial spots within the upper 2 quartiles of predictive values were selected for each of the 3 predictive models and compared to determine the level of overlap between each method (Fig. 3e). Spots in the upper 2 quartile predictive values in at least 2 separate methods were defined as positive for each cell type. The distribution of cells using our combined predictive pipeline showed strong mapping of MesLin cells to cortical bone (53%) and hematopoietic cells to the marrow compartment (83%) (Fig. 3f), consistent with the known cellular composition of these tissues. The presence of hematopoietic cells within the cortical (27%) and trabecular (59%) compartments likely reflects the fact that spatial spots designated as cortical or trabecular bone partially overlap with the neighboring marrow.
Fig. 2
Spatial spot deconvolution using scRNAseq. Overview of the deconvolution of spatial spots. First, scRNAseq data and spatial data are collected from the same or similar tissue. Second, scRNAseq data and spatial transcriptomics are then fed to three different deconvolution algorithms: Cell2Location, Seurat, and CellTrek. Cell2Location uses a Bayesian model to decompose the spatial expression count matrix into cell type signatures. Seurat employs bulk gene expression deconvolution based on a single-cell reference. CellTrek maps single cells to spatial locations. Finally, prediction results of the cell type abundance at each spatial spot are generated from the three algorithms
Fig. 3
Compositional deconvolution of spatial spots using scRNAseq. a UMAP of bone marrow scRNAseq datasets. b Probability that spatial spots contain MesLin cells. c Standard deviation (SD) of predictive values for each cell cluster within each spatial spot. Higher values denote greater variation in predictive values and a greater discernment of cell types within each spatial spot. d Pie chart showing the probability of each indicated cell type being assigned to each spatial region using the three indicated predictive packages. e Overlap between prediction methods (Seurat, CellTrek, and Cell2Location) of spatial spots proposed to contain MesLin cells. f Pie chart showing the probability of each indicated cell type being assigned to each spatial region using the combined predictive pipeline
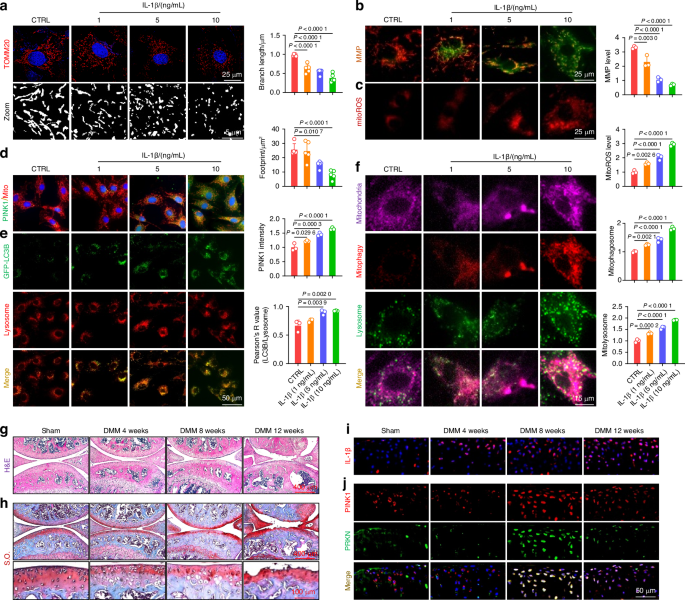
With this analytical pipeline, MesLin cells showed increased predictive probabilities within the cortical and, to a lesser extent, trabecular spatial spots (Fig. 4a, insert), likely corresponding to the high enrichment of osteoblasts and osteocytes within the bone. For confirmation of the use of these selection criteria, MesLin+ spatial spots were isolated from the marrow (Fig. 4a), and differentially expressed genes were calculated on MesLin+ marrow spots relative to MesLin− marrow spots. Pathway analysis of DEGs showed enrichment in terms linked to matrix production/organization, as well as skeletal development and ossification, within MesLin+ spots, while MesLin− spots were highly enriched for erythropoiesis, immune cells, and cell proliferation (Fig. 4b). To further refine this spatial localization of scRNAseq clusters, we next reanalyzed and subclustered MesLin cells (Fig. 4c). The expression pattern of marker genes (Fig. 4c and Table S1) revealed mature osteoblasts (OBs) and early osteocytes (Ocys), along with two previously proposed, heterogeneous SSPC populations, PαS cells, defined here as expressing high levels of Pdgfra and Ly6a (encoding SCA1) (PαS), and CXCL12-abundant reticular (CAR) cells, demarcated here by the expression of Cxcl12 and Lepr. To characterize the spatial distribution of these SSPC clusters, we applied a similar predictive modeling workflow as described above, with MesLin+ spatial spots further divided into these two proposed SSPC cell subtypes, along with mature osteoblasts to serve as a control (Fig. 4d). Quantitative analyses of subtype distribution suggest that PαS cells were highly enriched within the cortical bone, especially along the outer cortical surface consistent with the periosteum (Fig. 4d, e). In contrast, CAR cells were found primarily within the bone marrow, consistent with bone marrow SSPCs. Osteoblasts were associated primarily with trabecular spatial spots and to a much lesser extent within cortical spatial spots, consistent with the high bone remodeling frequently observed within trabecular compartments. Our combined predictive pipeline approach demonstrates the collective power of spatial transcriptomics and scRNAseq to overcome limitations in spatial resolution and spatially localize PαS and CAR cells to the periosteum and marrow, respectively.
Fig. 4
Proposed SSPC subtypes localize to spatially distinct skeletal regions. a Spatial spots identified as containing MesLin cells within the marrow. The insert shows the probability of MesLin cells localizing to each of the spatial regions. Dotted boxes denote the upper (red) and lower (blue) 2 quartiles. b Pathway analysis of marrow DEGs enriched in MesLin+ and MesLin− spatial spots. c UMAP of subclustered MesLin cells. d Predictive modeling of the indicated single-cell cluster onto spatial spots. e Predictive modeling quantification of the indicated MesLin subcluster onto segmented spatial data
Defining the cellular components of the marrow SSPC nicheHaving computationally derived the spatial locations of this heterogeneous Cxcl12+Lepr+ SSPC population within the bone marrow, we next investigated the other cellular components present within the bone marrow SSPC niche. We combined our predictive pipeline data for each cell cluster (Fig. 3f). Next, correlative analyses were conducted to determine the probability that SSPCs and each indicated cell type are present within the same spatial spots (i.e., which cell types are frequently found to be present within the same spatial location as SSPCs) (Fig. 5a). Correlations using all three predictive modeling packages (Seurat, CellTrek, Cell2Location) were used to quantify the slope of the spatial correlations between SSPCs and the cell types of interest (in log scale) and the significance of this correlation (Fig. 5b). These results suggest that Cxcl12+Lepr+ SSPCs within the bone marrow are most frequently spatially associated with smooth muscle cells, endothelial cells, macrophages, and, to a lesser extent, megakaryocytes. Conversely, SSPCs show a strong, negative spatial correlation with red blood cells (Fig. 5c). These results recapitulate previous works in the stem cell field,13 validating this approach to assess the cellular composition of the niche using spatial transcriptomics and our deconvolution pipeline.
Fig. 5
Identification of niche-resident cells. a Probability correlations indicating the likelihood that cell types are present within marrow Cxcl12+Lepr+ SSPC spatial spots. b Statistical analyses showing the likelihood that the indicated cell is present within the stem cell niche. Green values indicate significant positive enrichment; red values indicate significant negative enrichment. c Bubble plot showing cell types predicted to be within the niche. Cell types within the dotted boundary were significantly enriched. d UMAP of subclustered ECs. e Dot plot showing predictive values of where each EC subcluster maps to each spatial transcriptomic region. f Dot plot showing modular scoring of pathway activation within each EC subcluster. g Bubble plot showing EC subclusters predicted to be within the SSPC niche. h UMAP of subclustered Macs. i Dot plot showing predictive values of where each Mac subcluster maps to each spatial transcriptomic region. j Dot plot showing modular scoring of pathway activation within each Mac subcluster. k Bubble plot showing Mac subclusters predicted to be within the SSPC niche
To further refine the cellular constituents present within the bone marrow Cxcl12+Lepr+ SSPC niche, we subclustered and reanalyzed endothelial cell (Fig. 5d–g) and macrophage (Fig. 5h–k) single-cell populations for their association with SSPC-containing spatial spots. Endothelial cells were subclustered into 3 transcriptionally unique subpopulations (EC1-3) (Fig. 5d). EC1 was enriched for marker genes for sinusoidal vessels, while EC3 was enriched for arteriole markers4 (Fig. S3A). EC2 showed moderate expression for each vessel type but was enriched for angiogenic tip cells vs. stalk endothelial cells25 (Fig. S3B, C). These 3 endothelial cell subclusters exhibited differential mapping to our spatial data, with EC1 most closely associated with cortical bone surfaces, EC2 associated primarily with trabecular bone, and EC3 associated with the bone marrow and, to a lesser extent, trabecular bone (Fig. 5e). Pathway analysis of subcluster DEGs (Table S2) showed enriched activation of BMP signaling in EC1, TGFβ and hypoxia-related signaling in EC2, and Notch signaling in EC3 (Fig. 5f). Calculation of the correlation slopes revealed that EC3 was significantly and positively correlated with SSPC-containing bone marrow spatial spots, while the EC1 and EC2 subpopulations showed a strong negative and weak positive correlation, respectively (Figs. 5g and S2A). Similar subcluster analyses were conducted on macrophages (Mac1–4) (Fig. 5h), with spatial mapping showing distinct areas of enrichment for each of the 4 macrophage subclusters (Fig. 5i). Pathway analyses from subcluster DEGs (Table S3) showed unique enrichment in several pathways linked to cellular function and inflammatory status (Fig. 5j). Calculating the correlation slopes revealed that Mac2 was significantly and positively correlated with SSPC-containing bone marrow spatial spots (Figs. 5k and S2B). Mac3 cells showed a positive correlation with SSPC-containing spots similar to the total macrophage population but were not significantly enriched due to high variance in the correlative slopes between prediction methods (combined P value = 0.109 2), while both Mac1 and Mac4 associations were found to not be significant (Figs. 5k and S2B). These data demonstrate the feasibility of using single-cell prediction methods combined with spatial transcriptomics to identify heterogeneous populations of cell types frequently present within the stem cell niche. Furthermore, these results demonstrate that these cell types can be segmented by gene expression profiles and spatially subdivided to quantify enrichment within the SSPC niche.
Dissecting cell‒cell interactions within the SSPC nicheHaving established the cell types frequently present within the SSPC niche, we next sought to determine the signaling mechanisms occurring within the stem cell microenvironment. Differential gene expression analysis was conducted on SSPC+ spatial spots within the marrow relative to other marrow spots using each of the 3 methods of single-cell predictions (Table S4) or our combined method approach (Table S5). Pathway analysis of genes enriched within SSPC-containing marrow spots revealed enriched expression in several well-established morphogenetic pathways, including WNT, Notch, and TGFβ signaling (Fig. 6a). Next, we sought to use ligand‒receptor interactions to understand how these and other signaling cascades are regulated within the SSPC niche. Single-cell analysis packages have been previously developed to identify potential cell‒cell interaction mechanisms.26,27,28 However, they lack the spatial information to determine whether the proposed cell types both exist within a sufficiently close proximity and express the proposed ligand and receptor within this region. As such, it is challenging to distinguish computationally predicted (Fig. 6b, dotted arrows) from biologically relevant (Fig. 6b, solid arrows) signaling mechanisms. To overcome these limitations, we combined single-cell data with our spatial data to refine our list of total spatial and single-cell cluster DEGs into those genes that expressed known ligands or receptors, which were predicted to interact between cell types by CellChat, were mechanisms of cell‒cell interaction between the cell types predicted to be present within the SSPC niche and finally showed enriched expression within our predicted SSPC+ niche spatial spots (Fig. 6c and Table S6). Although unable to distinguish finer spatial organization, this combinatorial approach of scRNAseq and spatial transcriptomics can identify genes enriched within a 55 µm area (size of each spatial spot) around the predicted SSPC location. Probing of these different categories of overlapping DEGs allowed us to identify (i) niche signaling factors spatially restricted to the niche but showing widespread expression across multiple cell types (spatially restricted), (ii) genes that were enriched within cell types but not spatially restricted to the niche (cell type restricted), and (iii) genes that were found to show both cell type and spatially restricted expression (spatial and cell type restricted) (Fig. 6d). Similar results were obtained using RNAscope for Cd44-, Cd74-, and Ryr1-expressing cells surrounding Lepr+ SSPCs (Fig. 6e). Quantification of these in vivo results shows that while Cd44+ and, to a lesser extent, Ryr1+ cells are spatially restricted to within a few µm of Lepr+ SSPCs, Cd74+ cells were more ubiquitously dispersed (Fig. 6f). These data suggest that combining spatial and single-cell analyses can localize potential ligand‒receptor cell‒cell interactions predicted by scRNAseq algorithms within SSPC niche signaling in an unbiased manner to reveal new components of niche biology.
Fig. 6
Signaling within the stem cell niche using spatial transcriptomics. a Pathway analysis of DEGs enriched with niche spatial spots relative to other spatial spots within the bone marrow. b Schematic of cell‒cell interactions. While scRNAseq can be used to predict ligand/receptor pairs between cell types, the addition of spatial information can distinguish biologically relevant interactions occurring between cells with close proximity (within circle, solid arrows) from those occurring between distant cells unlikely to occur in vivo (outside of circle, dotted arrows). c Predicted cell‒cell interactions using single-cell spatial transcriptomics. Genes were defined as showing enriched expression within select cell types (brown), encoding known receptors or ligands (yellow), predicted to communicate using CellChat (green), and being expressed by those cell types present within the stem cell niche (magenta). Genes were overlaid with spatial DEGs (blue). d Sample genes identified from cell‒cell interaction found to be spatially restricted to the niche but expressed by numerous different cell types (Cd44), genes expressed by only select cell type but present throughout the marrow (Cd74), and genes restricted both spatially and by cell type (Ryr1). e RNAscope of bone marrow showing Lepr-expressing SSPCs (red) and Cd44 (left), Cd74 (middle) and Ryr1 (right) (green). Bar, 10 µm. f Distribution of distances between the indicated niche cell markers and Lepr+ SSPCs. Numbers above the graph represent the average±SD of the distance between cells. n = 50 cells per marker. aP < 0.001 vs. Cd74, bP < 0.001 vs. Ryr1
Signal gradients establish domains within the bone marrowIn addition to local signaling axes present within the SSPC niche, we further hypothesized that broader signaling gradients exist within the bone marrow that establish domains within the marrow cavity. These microdomains would be the result of secreted ligands or the availability of nutrients and oxygen and act in a coordinated fashion, likely affecting all cells based on their proximity to major tissue areas such as the bone surface or blood vessels. To assess these gradients in an unbiased fashion, we conducted spatial-time analyses on marrow spatial spots (Fig. 7). This technique utilizes a manually designated reference line and then measures the distance between each spatial spot and the nearest point along this reference surface.24 First, spatial spots were aligned based on their relative proximity to the nearest blood vessel, trabecular bone, or cortical bone surface (Fig. 7a). Genes whose expression fluctuated as a function of their proximity to their reference surface were then identified (Fig. 7b). Pathway analysis was then used to identify major regulatory networks altered relative to their distance from the reference tissue.
Fig. 7
Spatial-time analysis reveals gradients of signals originating from the blood vessels. a Spatial distance calculations showing the minimal distance of each marrow spatial spot to the nearest vessel (left), trabecular bone surface (middle), and cortical bone surface (right). b Identification of genes that are differentially regulated across the marrow as a function of their distance to the nearest blood vessel. c Module scoring showing activation of the indicated metabolic pathways within marrow spatial spots relative to their distance to the nearest blood vessel (left), trabecular (middle), or cortical (right) surface. d Module scoring showing activation of the indicated morphogenetic pathways within marrow spatial spots relative to their distance to the nearest blood vessel (left), trabecular (middle), or cortical (right) surface. e Immunofluorescence staining of TGFβ activation (p-SMAD3) and PDGFRα in mouse long bone. Blood vessels are denoted by endomucin (green). Dotted lines denote the bone boundary. Bar, 100 µm
To observe overall pathway activation across SpatialTime as a function of the distance from the nearest vessel, trabecular, or cortical bone surface, we conducted module scoring, which calculates the average expression of a gene list curated from known KEGG pathways relative to background. Scoring for genes linked to bioenergetics, we observed high levels of glycolytic gene expression immediately adjacent to blood vessels, immediately adjacent and distant from the trabecular surface, and distant from the cortical bone surface (Fig. 7c). In contrast, genes linked to oxidative phosphorylation (OxPhos) were predominantly found within spatial marrow spots distant from blood vessels and intermediate from both trabecular and cortical bone surfaces. Finally, fatty acid (FA) metabolism was found to be predominantly associated with the cortical bone surface and at intermediate distances from trabecular bone surfaces (Fig. 7c). In addition to metabolic regulation, pathway analysis identified changes in gradients of signal activation associated with major morphogenetic pathways (Fig. 7d). Platelet-derived growth factor (PDGF) signaling was found to be highly active both adjacent to and distant from blood vessels, as well as adjacent to trabecular bone surfaces. In contrast, WNT signaling was high immediately adjacent to blood vessels and distant from both trabecular and bone surfaces, while WNT signaling appeared conversely low distant to vessels and adjacent to trabecular and cortical bone surfaces (Fig. 7d). Both bone morphogenetic protein (BMP) and transforming growth factor beta (TGFβ) signaling were preferentially high near trabecular and cortical surfaces and declined at more distant marrow spots (Fig. 7d). Histological validation confirmed the preferential activation of TGFβ signaling (denoted by p-SMAD3 staining) near cortical and trabecular surfaces, as well as preferential staining for PDGF receptor alpha (PDGFRα) near trabecular vascular surfaces (Fig. 7e). Overall, these unbiased analyses identify microdomains of signaling gradients, cellular function, and metabolism in response to the distance from blood vessels and bone surfaces.
Comments (0)