Animals
Before animal usage, the protocol was first reviewed and approved by the IRB at the Institute of Biomedical Engineering, West China School of Basic Medical Sciences & Forensic Medicine (K2021015). Female C57BL/6 mice (6 weeks, 18 ~ 22 g) were purchased from Chengdu Dossy Biological Technology Co., Ltd. (Chengdu, China). The mice were housed under specific pathogen-free conditions with a constant temperature of 25 °C and a 12 h light/dark cycle. The cages were regularly cleaned and supplied with plenty of food and water. The mice were fed an alfalfa-free diet for at least 1 week before the experiment.
OVX mouse model
The female C57BL/6 mice were randomly divided into three groups: sham group (sham), OVX group (OVX), and AnxA5-treated OVX group (OVX + AnxA5). For the OVX group, ovariectomy was performed as previously described.41 Briefly, the mouse was anesthetized with 1.5% isoflurane (R510-22, RWD Life Science Co., Ltd., Shenzhen, China) at a rate of 0.5–1.0 L·min−1 and then placed on a 37 °C thermostatic heating plate in a prone position. After the hair was shaved and the dorsal area was sterilized with 75% alcohol, an approximately 1 cm cut was made in the middle dorsal skin, and the ovary was thoroughly excised with scissors. The wound was sutured and wiped with 2% iodophor disinfectant. The bilateral ovaries were both removed as described above. For the sham group, the same procedure was conducted without removing the ovaries. For the OVX + AnxA5 group, one month after the ovariectomy surgery, the mice were intravenously injected with 1 mg·kg−1 AnxA5 twice a week for 8 weeks. The mice in each group were sacrificed 3 months after the surgery. The serum and bilateral femurs and tibias were collected for subsequent in vivo and in vitro analyses.
Cell isolation and culture
The preosteoblast cell line MC3T3-E1 (Jennio Biotechnology Co., Ltd., Guangzhou, China) was cultured with alpha minimal essential media (α-MEM, Gibco, California, USA) with 10% fetal bovine serum (FBS, Aoke, Chengdu, China) and 1% penicillin‒streptomycin solution in a humidified atmosphere of 5% CO2 at 37 °C.
C57BL/6 mice were used in this study to isolate primary osteoblasts and BMSCs. BMSCs were isolated from the bone marrow of the femurs and tibias according to the protocol provided.83 Briefly, mice were anesthetized and killed, and the long bones were immediately dissected under sterile conditions and placed in α-MEM with 1% penicillin‒streptomycin (PS, SV30010, HyClone, Utah, USA). The soft tissue and periosteum attached to the bone were thoroughly removed, and the epiphyses were cut. The marrow in the bone cavities was then gently flushed out with α-MEM with 1% PS. After centrifugation at 1 500 × g for 5 min, the marrow pellets were resuspended in fresh complete α-MEM and transferred to cell culture flasks. The media were replaced to remove nonadherent cells after 3 h, 24 h and 48 h of culture. The remaining adherent cells, BMSCs, were cultured until reaching 90% confluence. Cells at passages 2–4 were used in the experiment. The above marrow-depleted bones were then used for osteoblast isolation as described previously.84 Briefly, the bones were excised into approximately 1–2 mm3 small pieces and digested with 0.05% trypsin-EDTA (25200056, HyClone, Utah, USA) for 20 min, followed by 3 washes with phosphate-buffered saline (PBS, SH30256, HyClone, Utah, USA). Subsequently, collagenase IA (1 mg·mL−1; C8140, Solarbio, Beijing, China) digestion was performed for 60 min and repeated twice. The collagenase solution was collected each time to obtain osteoblasts by centrifuging at 1 000 r·min−1 for 5 min. The isolated osteoblasts were then cultured with fresh complete α-MEM media at 37 °C in a 5% CO2 incubator. For purification, the digested cells were cultured for 20 min before transferring the nonadherent cells to a new culture flask. Passages 3–5 were used for the follow-up experiment.
Antibodies and reagents
Detailed information about the antibodies used for Western blotting analysis and immunofluorescence staining is shown in Table S1. Rapamycin (Rap, 100 nmol·L−1; V900930, Sigma-Aldrich, Missouri, USA) was used to induce autophagy by inhibiting the activity of mTOR in this study. Osteogenic induction media (OM) was prepared by adding additional ascorbic acid (100 μmol·L−1; A7506, Sigma-Aldrich, Missouri, USA), dexamethasone (10 nmol·L−1; D4902, Sigma-Aldrich, Missouri, USA) and β-glycerophosphate (10 mmol·L−1; G9422, Sigma-Aldrich, Missouri, USA) to the complete α-MEM media.28
Plasmid and transfection
For AnxA5 knockdown, MC3T3-E1 cells/primary osteoblasts were transfected with the shRNA plasmid (Tsingke Biotechnology Co., Ltd., Beijing, China) targeting AnxA5 toward the sequence of 5ʹ-CCGGCGTGAAGTCTATTCGGAGCATCTCGAGATGCTCCGAATAGACTTCACGTTTTTG-3ʹ using Lipofectamine 8000 (Beyotime, China) according to the manufacturer’s protocol (named the shAnxA5 group). The shRNA plasmid with the sequence 5ʹ-CCGGGGTTCTCCGAACGTGTCACGTCTCGAGACGTGACACGTTCGGAGAACCTTTTTGAATT-3ʹ served as the negative control (named the shControl group). After 48 h of incubation, complete medium containing 5 μg·mL−1 puromycin (ST551, Beyotime, Shanghai, China) was used to select shAnxA5 stably transfected cells. For AnxA5 overexpression, the plasmid was constructed by Shanghai GenePharma Co., Ltd. (named the AnxA5 overexpression group). The vector plasmid served as the negative control (named the vector group). MC3T3-E1 cells or osteoblasts were transfected with the plasmids using Lipofectamine 8000 (C0533, Beyotime, Shanghai, China) according to the manufacturer’s protocol. After 48 h of transfection, G418 (500 μg·mL−1; ST081, Beyotime, Shanghai, China) was used to select stably transfected cells. Once the cells reached confluency, they were collected for final transfection efficiency identification by Western blotting analysis.
Isolation of matrix vesicles
For bone-derived MVs, femurs and tibias were dissected, and the attached muscles and periosteum were cleaned. Marrow-depleted bones were then crushed to powder in liquid nitrogen, followed by digestion with collagenase IA (100 U·mL−1, in Hank’s balanced salt solution) for 3 h in a shaking incubator at 37 °C with a shaking speed of 200 r·min−1. The solution was then collected and filtered with a 0.48 μm filter. The MVs were finally purified using an exosome isolation kit (EX010, Shanghai Gefan Biotechnology Co., Ltd., Shanghai, China) according to the manufacturer’s protocol.
For cell-derived MVs, once the cell confluency reached approximately 80%, the growth media were replaced with α-MEM with 10% MV-free FBS and 1% PS. The matrix vesicles from the ECM were isolated after 48 h of incubation according to the protocol previously described.33 Briefly, the growth media were removed, and the remaining cells were washed 3 times with PBS. Collagenase IA (0.8 mg·mL−1; C8140, Solarbio, Beijing, China) was then used to digest the ECM and release the MVs. After 4 h of incubation at 37 °C, the digestion solution was collected and centrifuged as follows: 300 × g for 20 min (to discard pelleted cells), 3 000 × g for 20 min (to discard pelleted cell debris), 10 000 × g for 30 min (to discard pelleted apoptotic bodies and microvesicles), and 100 000 × g for 120 min to obtain the MV pellet. Finally, the pellet was resuspended in 100 µL of PBS and either used immediately or stored at −80 °C.
Protein extraction, quantification and Western blotting
Collected cells, MVs or preground bone tissues were lysed with RIPA lysis buffer with protease inhibitor, phosphatase inhibitor and phenylmethylsulfonyl fluoride for 30 min on ice. The protein was extracted by centrifugation at 14 000 × g for 10 min at 4 °C, followed by determination of the protein concentration using a BCA protein assay kit (P0012, Beyotime, Shanghai, China). Western blot assays were performed as previously described.85 Equal amounts of protein (20 µg) from each sample were subjected to electrophoresis on 8%/10%/12% sodium dodecyl sulfate‒polyacrylamide gels and then transferred to a polyvinylidene difluoride (PVDF) membrane. After the membrane was blocked with 5% skim milk at room temperature for 2 h, it was incubated overnight with specific primary antibodies (diluted in 5% skim milk) on a roller bank at 4 °C and treated with the corresponding HRP-conjugated secondary antibodies for 1–2 h at room temperature. The results were finally visualized and analyzed with enhanced chemiluminescence using a Molecular Image® Chemi-DocTM XRS+ system with Image LabTM Software. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as the internal control in this study.
Immunofluorescence of cells/tissues
For the immunofluorescence of cells, 1 × 105 cells of each group were seeded onto a coverslip placed in 24-well plates and cultured with osteogenic induction media for 7 days. The cells were then rinsed three times with PBS and fixed with 4% paraformaldehyde for 15 min. After the cells were blocked with 5% bovine serum albumin (BSA, BS114, Biosharp, Hefei, China) for 30 min at room temperature, they were incubated with primary antibodies (1:500, diluted in 5% BSA) overnight at 4 °C. The corresponding fluorochrome-labeled secondary antibodies (1:1 000, diluted in 1% BSA) were applied to the cells for 60 min after washing with PBS three times. The nuclei were subsequently stained with 4′, 6-diamidino-2-phenylindole (DAPI, 1:800, C1002, Beyotime, Shanghai, China) for 10 min and rinsed five times with PBS gently. The images were captured using a Zeiss (LSM710, Oberkochen, Germany) confocal microscope.
For the immunofluorescence of bone tissues, the paraffin sections were deparaffinized in xylene and hydrated in graded ethanol, followed by immersion in heated sodium citrate buffer (10 mmol·L–1, pH 6.0) for antigen retrieval. Subsequently, the samples were blocked, incubated with antibodies and stained for nuclei as described above.
Coimmunoprecipitation (co-IP) of AnxA5
The coimmunoprecipitation (co-IP) assay was performed as described previously.86 Briefly, 1 mg of total protein from the lysates was incubated with 1 μL of primary antibodies against AnxA5 overnight at 4 °C. Then, 20 µL of A/G agarose beads (P2012, Beyotime, Shanghai, China) was added to the above solution for 2 h of incubation with gentle vibration at 4 °C. After three washes with RIPA solution, the beads were precipitated by centrifuging at 5 000 × g for 1 min at 4 °C. The protein was then eluted with 2× loading buffer by boiling for 8 min. The following immunoblotting assay was similar to the protocol of Western blotting described above.
Proteomic analysis
MVs (1 mg in PBS) secreted by normal or osteoporotic individuals were purified as described above and were then used for LC–MS/MS analysis. The analysis was processed by KangChen Biotech Co., Ltd. The GO analysis was performed based on differentially expressed proteins.
Alizarin red staining
The cells reaching confluency were cultured with osteogenic induction media for 7 days or 14 days and were then fixed with 95% alcohol for 15 min. Next, the cells were washed three times and stained with 2% Alizarin red solution (pH 4.2, C0138, Beyotime, Shanghai, China) for 30 min at room temperature. After the excess dye was removed and the cells were rinsed five times with ddH2O, the mineralized nodules were observed by an inverted microscope (CK2, Olympus, Tokyo, Japan).
Alkaline phosphatase staining
The cells reaching confluency were cultured with osteogenic induction media for 7 days. ALP staining was performed using a kit (C3206, Beyotime, Shanghai, China) according to the manufacturer’s protocol. Briefly, cells were fixed with 4% paraformaldehyde for 15 min and subsequently coincubated with prepared working solution for 0.5–1 h at room temperature. The reaction was terminated by rinsing the cells 3 times with ddH2O.
Alkaline phosphatase activity assay
The cells were cultured with osteogenic media for 7 days, lysed with RIPA lysis buffer without the addition of phosphatase inhibitor on ice and centrifuged at 14 000 r·min−1 for 10 min. ALP activity in the supernatant was measured using p-nitrophenol phosphate substrate supplied in an ALP assay kit (P0321S, Beyotime, Shanghai, China). The absorbance was measured at 405 nm, and the final ALP activity was normalized to the protein concentration.
Nanoparticle tracking analysis
The MV pellet was resuspended in 100 μL of PBS, and the size and concentration were subsequently measured by nanoparticle tracking analysis (NTA) using a ZetaView instrument (PMX120, Particle Metrix, Munich, Germany). Data acquisition and analysis were performed using Zetaview Analytical Software.
Transmission electron microscopy (TEM)
For observation of the morphology of MVs, the samples resuspended in PBS were dropped on a copper mesh for 2 min of adsorption. When the solution was removed using filter papers, 2% phosphotungstic acid solution was applied to the copper grid for 10 min at room temperature to perform negative staining. After the staining solution was removed and the sample was dried, the MVs were observed and captured using TEM (HT7800, Hitachi High-Tec Co., Ltd., Tokyo, Japan) at 80 kV.
For observation of the MVs deposited in bone, the dissected distal femurs were fixed in 2.5% glutaraldehyde for 12 h and decalcified thoroughly with EDTA solution (G1107, Servicebio, Wuhan, China). After three rinses with PBS, the tissues underwent gradient acetone dehydration, 100% acetone-Epon 812 embedding, and ultrathin sectioning in sequence. The samples were then double‐stained with uranyl acetate-lead citrate, and the MVs were detected by TEM.
For determination of the formation of autophagosomes, the confluent cells in each group were collected and centrifuged at 1 500 r·min−1 for 5 min. Then, the cell pellets were fixed, dehydrated, embedded and sliced as described above. The autophagosomes were observed by TEM.
Scanning electron microscopy (SEM)
For observation of the MVs adhered to the ECM, the cells were fixed with glutaraldehyde fixative, especially for electron microscopy (2.5%), overnight at 4 °C after 7 days of osteogenic induction culture. After three rinses with ddH2O, the samples were dehydrated using graded ethanol (30%, 50%, 70%, 85%, 95%, 100%, 100%) for 15 min each time. The dehydrated samples were then placed in a critical point dryer for CO2 drying, sputtered with gold and observed with SEM (Phenom Prox, Phenom-World, Eindhoven, Netherlands).
MV uptake and coculture assays
BMSCs were seeded on 10 mm glass coverslips placed in 24-well plates and cultured overnight. Equal amounts of purified MVs (1 × 1010) were labeled with DiI (C1036, Beyotime, Shanghai, China) according to the manufacturer’s instructions and added to the growth media in each well. After 24 h of coincubation, the uptake of MVs by BMSCs was analyzed by confocal microscopy. The nuclei were stained with DAPI after 15 min of fixation in 4% paraformaldehyde.
For determination of the effect of MVs on the osteogenic differentiation of BMSCs, 5 × 105 cells per well BMSCs were seeded into 6-well plates and cultured overnight. The cells were then treated with fresh growth medium containing equal concentrations of MVs, which was replaced every other day. Total protein was collected for further Western blotting analysis after 5 days of coincubation.
In vitro MV-ECM/collagen I adhesion assay
For the in vitro MV-ECM adhesion assay, ECM was prepared on coverslips placed in 24-well plates. MC3T3-E1 cells were seeded on coverslips, cultured to confluence, and then incubated with osteogenic induction media for 7 days. The cells were then rinsed three times with PBS and decellularized by treatment with 20 mmol·L−1 NH4OH/0.25% Triton X-100 at room temperature for 3 min. The cell debris was removed, and the remaining decellularized ECM was washed three times with PBS for subsequent use. Equal amounts of DiI-labeled MVs (1 × 1010) were diluted in complete growth media and added to the prepared decellularized ECM. After 72 h of incubation at 37 °C, the anchored MVs on the ECM were detected by fluorescence microscopy (Olympus, Tokyo, Japan).
For the MV-collagen I adhesion assay, glass coverslips in 24-well plates were coated with type I collagen at a concentration of 2 mg·mL−1 with 0.006 mol·L−1 acetic acid and 0.1 mol·L−1 NaOH and placed at room temperature for 4 h. Equal amounts of DiI-labeled MVs were then added to the collagen I-coated coverslips and cocultured for 72 h. Collagen I was then determined using the IF assay described above, and the MVs adherent to the ECM were also observed by confocal microscopy.
Micro-CT analysis
The dissected tibias and femurs were fixed with 4% paraformaldehyde for 24 h at room temperature and subsequently scanned ex vivo using a micro-CT scanner (mCT50, Scanco Medical, Bassersdorf, Switzerland). Three-dimensional reconstructions of tibias and femurs were performed with a micro-CT system. The bone morphometric parameters, including bone mineral density (BMD), ratio of bone volume to tissue volume (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th) and trabecular separation (Tb.Sp), were analyzed.
Histological staining and analysis
The isolated tibias and femurs were fixed in 4% paraformaldehyde for 24 h and then decalcified with EDTA solution in a constant rotating shaker at room temperature. After dehydration with gradient ethanol, the bone tissues were embedded in paraffin and cut into 5–7 μm sections. The sectioned tissues were placed on slides and dried overnight for subsequent histological analysis, including hematoxylin and eosin staining (H&E), Goldner staining and TRAP staining. The stained tissues were scanned using a digital slice scanner (VS200, Olympus, Tokyo, Japan), and the images were captured using CaseViewer Software.
Biophotonic imaging analysis
DiI-labeled MVs (100 μg in PBS) were intravenously injected into C57BL/6 mice via the tail. After 12 h, the bilateral tibias and femurs were extracted to be detected by an IVIS spectrum imaging system (µCT45, Scanco Medical, Bassersdorf, Switzerland). The deposition of MVs in bones was evaluated by average radiant efficiency.
Statistical analysis
The results are presented as the mean ± standard deviation (SD). All data were analyzed with GraphPad Prism 9 software. The statistical significance was analyzed by two-tailed Student’s t test or one-way analysis of variance (ANOVA) with Tukey’s post hoc tests. P < 0.05 was considered statistically significant.

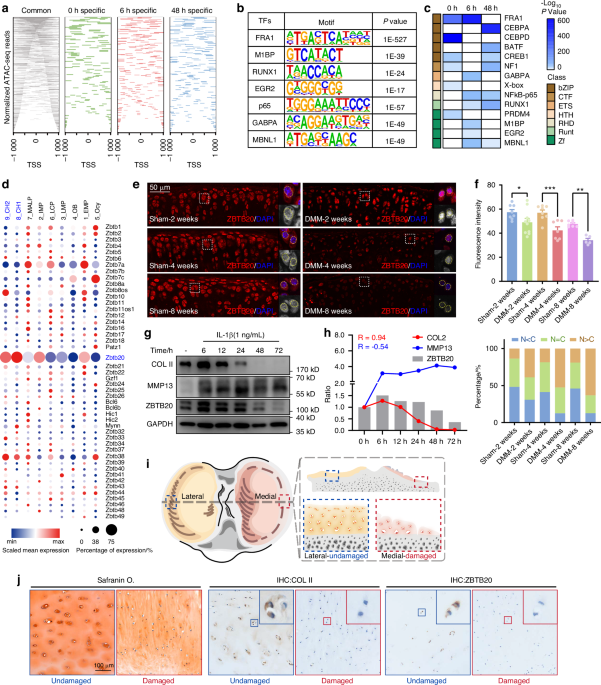
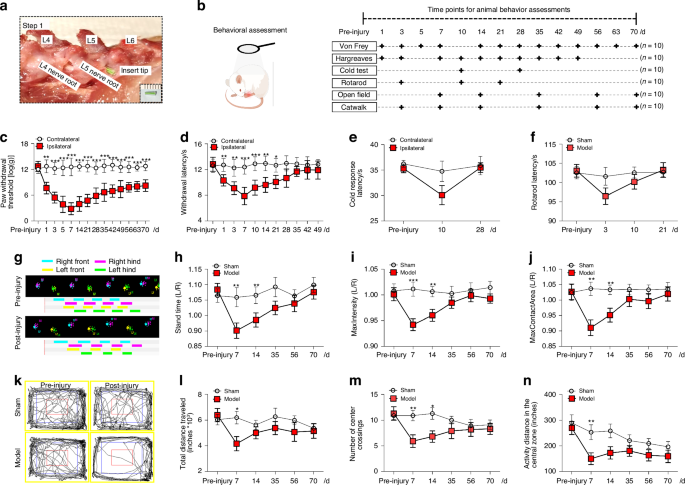
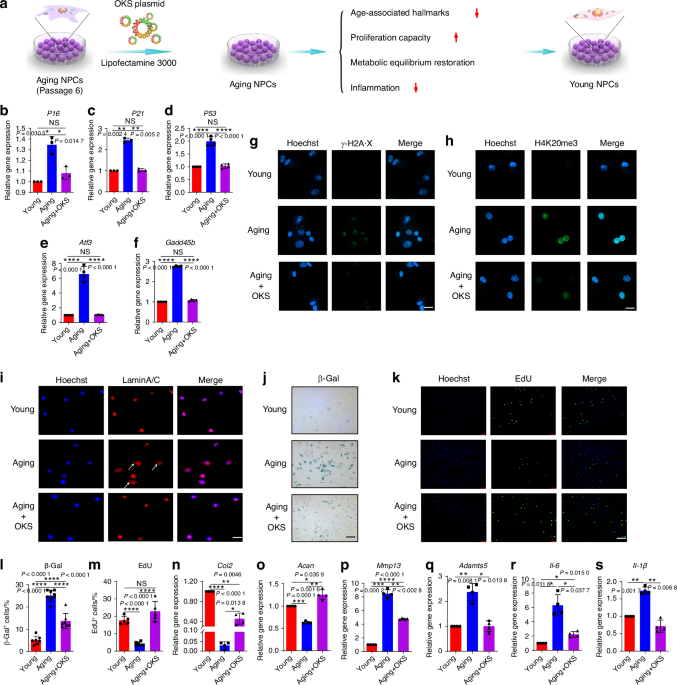
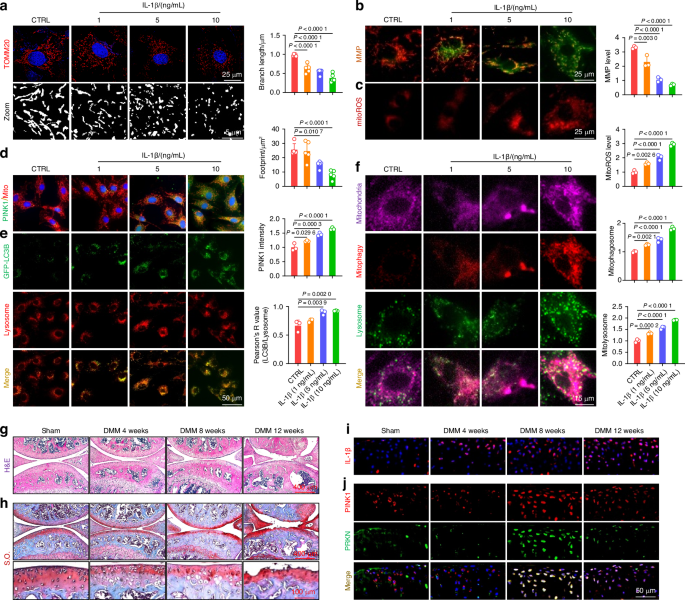

Comments (0)