
Remember me






Inborn errors of immunity (IEI) are a heterogeneous group of monogenetic disorders that affect the innate or adaptive immune system. Clinical manifestations may include severe, atypical or recurrent infections as well as immune dysregulation characterized by autoimmunity, autoinflammation, atopy, and increased risk of cancer. To date, approximately 500 IEI causing genes have been described, and with the widely available sequencing methods this number continues to grow rapidly [1]. Established treatment options consist of immune modulatory drugs, antimicrobial prophylaxis, immunoglobulin replacement therapy and, for severe IEI, allogeneic hematopoietic stem cell transplantation (HSCT). With HSCT, a patient's hematopoietic stem cells (HSCs) are replaced by healthy donor HSCs. The first successful HSCT were performed in 1968, to treat severe combined immunodeficiency (SCID) [2,3]. HSCT emerged as a life-saving therapy, but often resulted in partial or insufficient cure, with high morbidity and mortality, particularly in the large subgroup of patients dependent on treatment with a mismatched or haploidentical donor. Unfavorable HSCT outcomes with mismatched donors, growing knowledge on the genetic causes of SCID and the recognition of revertants as naturally occurring phenomenon leading to correction of the disease-causing genetic defect [4,5] together prompted the development of gene therapy in the 1990s. Gene therapy aims to correct a patient's own HSCs, thereby avoiding any risk of alloreactivity related complications [6]. X-linked SCID (X-SCID) and adenosine deaminase-deficient severe combined immunodeficiency (ADA-SCID) were the first genetic diseases to be successfully treated by gene therapy [7–13] eventually resulting in Strimvelis as the first, and to date only, ex vivo gene therapy with market authorization for the treatment of ADA-SCID [14]. In the past decades new viral vectors with a more favorable safety profile have been developed, integration site analysis techniques for safety monitoring have improved and conditioning is increasingly being used to facilitate engraftment of the corrected cells. Still, several challenges remain, most importantly the efficacy, safety and access to gene therapy.
In parallel with the development of gene therapy, the field of HSCT has made great strides since the 1990s, with improved human leukocyte antigen (HLA) typing and donor selection strategies, more effective and less toxic conditioning regimens and innovation in stem cell graft manipulation. Advances were also made in the management of toxicities, graft versus host disease (GvHD) and infections. However, HSCT for IEI still comes with a significant risk of morbidity and mortality in up to 25% of the patients, particularly in those patients transplanted with a mismatched donor [15–21].
This review will summarize the main aspects of gene therapy in IEI, the progress made and the remaining challenges.
 Box 1:
Box 1: no caption available
GENE THERAPYCurrently, all clinically tested gene therapy strategies for IEI rely on gene addition using viral gene transfer vectors (Table 1). Newer methods, based on gene editing with engineered nucleases, are still at a preclinical stage. In the next section, the two different approaches will be explained, with a focus on viral gene addition (Fig. 1). Importantly, regardless of the strategy, the autologous HSCs need to be collected, purified and manipulated ex vivo in optimal culture conditions to maintain their stemness characteristics [22]. Just as for HSCT, autologous HSCs can be obtained from peripheral blood by leukapheresis or from bone marrow by aspiration. Prior to leukapheresis patients are generally treated with a combination of GCS-F and plerixafor (CXCR4 antagonist) to mobilize their HSCs from bone marrow to peripheral blood [23]. The mobilized HSC approach is increasingly applied as this results in a much higher HSC yield which has a positive impact on hematological and immunological recovery [24]. Although initial SCID gene therapy studies were performed without conditioning it has become clear, similar to what is known for regular HSCT, that a certain level of myeloablative conditioning is required for successful and sustainable engraftment of corrected cells [8,25–28]. The amount and type of conditioning needed depends on the underlying disease and the selective advantage of the corrected cells [29].
Table 1 - Gene therapies using viral gene addition Disease name Gene Vector Status Ref/trial ADA-SCID ADA gRV Commercial product in Europe (strimvelis) [14] ADA LV Compassionate use (GOSH)/Human trial (UCLA/NIH) [79▪▪] X-SCID IL2RG SIN gRV Human trial completed [37] IL2RG LV Human trial GOSH/BCH/UCL open [82,83]ADA-SCID, adenosine deaminase-deficient severe combined immunodeficiency (SCID); CGD, chronic granulomatous disease; fHLH, familial hemophagocytic lymphohistiocytosis; gRV, gamma-retroviral; IPEX, immune dysregulation, polyendocrinopathy enteropathy X-linked; LAD, leukocyte adhesion deficiency; LV, lentiviral; RAG, recombination activating gene; WAS, Wiskott-Aldrich syndrome; XIAP, X-linked inhibitor of apoptosis protein deficiency; XLA, X-linked agammaglobulinemia; XLP, X-linked lymphoproliferative disease; X-SCID, X-linked SCID.
 FIGURE 1:
FIGURE 1: Different approaches to gene therapy for IEI. In all types of gene therapy for IEI, ex vivo gene therapy is used, in which a viral vector is used to genetically modify cells from the patients with therapeutic intend. Cells could include T cells, HSCs or other cell types. For gene addition (left), gRV or LV vectors are used to add the therapeutic gene to the genome of the target cells. This occurs semi-randomly, with slight preferences for integration sites depending on the vector used. For gene editing, a specific site can be targeted to change the DNA and correct the mutation. This can be accomplished by using an engineered nuclease such as CRISPR/Cas to introduce a single or double stranded DNA break in combination with a repair template. This strategy can be used to add a full cDNA in the same gene under control of the endogenous regulatory elements. Alternatively, it can be used to add a full cDNA including regulatory elements into a known safe region of the genome elsewhere (safe harbor). This targeted approach avoids insertional mutagenesis, but allows gene expression to be controlled by design. gRV, gamma-retroviruses; HSC, hematopoietic stem cell; IEI, inborn errors of immunity; LV, lentiviral.
Viral gene additionVirus based vectors make use of the ability of RNA viruses to infect and stably integrate genetic material into the target cell's genome. These vectors contain a correct copy of the therapeutic gene (transgene) along with regulatory elements that control gene expression, such as promoters and enhancers (Fig. 1). The genes necessary for viral replication have been removed from these vectors, while maintaining features required for gene transfer, such as the LTR (long terminal repeat). This way, viral vectors can infect and stably transfer a therapeutic transgene into the patient HSC's genome (transduction). In general, the insertion of the transgene happens semi-randomly throughout the host's genome, but different viruses have different preferences thereby giving rise to a different integration pattern [30]. The number of viral vector transgenes integrated in the genome per cell is referred to as vector copy number (VCN) [31]. This is usually determined at population level and not at a single cell basis. The required therapeutic protein level depends on the disease and is determined by both VCN and strength of the regulatory elements of the transgene.
The first clinical trials of gene therapy used gamma-retroviruses (gRV) and successful T cell reconstitution was achieved for X-SCID [7,12], ADA [8,11] and WAS [32,33]. Unfortunately, several patients developed hematological malignancies due to preferential integration near transcription initiation sites of other genes (including oncogenes). The strong enhancer activity of the viral LTR in a number of cases led to increased and deregulated expression of the nearby oncogenes [33–36]. Newer generations of self-inactivating (SIN) gRV vectors have been developed with deletion of enhancer elements of the LTR, which resulted in better safety [37]. In recent years, clinical trials have predominantly used SIN-lentiviral (LV) vectors [38], with LTRs without enhancer and weaker or lineage-specific promoters. In addition, the insertional pattern of LV vectors seems safer than that of gRV vectors, because LV have no insertional preference for transcription initiation sites [30]. Lastly, LV transduction also works on quiescent HSCs enabling retention of stemness [38].
Safety assays to test for genotoxicity of viral vectors are paramount in both the preclinical and clinical studies, and several are currently being used [39]. In the most commonly used In Vitro Immortalization (IVIM) assay, HSCs are expanded after transduction with retroviral vectors. Following limiting dilution, nonimmortalized cells stop proliferating, whereas insertional mutants give rise to clonal outgrowth. With the surrogate assay for genotoxicity assessment (SAGA), the gene expression profile of transduced murine HSCs is analyzed and machine learning is used to detect dysregulated genes as immortalization signature. In addition, advanced integration site analysis using next generation sequencing enables researchers and clinicians to monitor patients enrolled in clinical trials closely and detect any preferential vector integration pattern leading to clonal outgrowth at a very early stage. To date, no hematological malignancies have been reported in patients with IEI treated with LV vector gene therapy, but the risk still remains that insertional mutagenic events occur.
Gene editingOver the last decade the field of gene editing has grown exponentially [40]. This technique introduces a single (nick) or double strand DNA break (DSB) at a specific target site in the DNA and relies on the natural process of homology directed repair to introduce a template sequence (donor template or repair template). These nicks or breaks are made using engineered nucleases such as zinc-finger nuclease (ZFN), transcription activator-like effector nucleases (TALEN) and CRISPR-Cas. Gene editing can be used to correct a specific mutation at the endogenous locus, which has been done in vitro for X-CGD [41,42] and RAG2 [43,44] or to add the therapeutic gene to the same locus under the control of its endogenous promoter. Proof of principle of this approach has been demonstrated for X-SCID [45–47], X-CGD [48], CD40L [49▪,50,51], CTLA4 [52], XLP [53], RAG1 [54,55] and Wiskott–Aldrich syndrome (WAS) [56]. With both approaches, endogenous gene regulation is kept intact resulting in physiological expression. This is crucial when transcriptional control of the gene is tightly regulated, as in the case of CD40L, where constitutive expression leads to lymphoproliferative disorders [57,58].
Alternatively, a correct copy plus gene regulation elements can be added to a well studied safe harbor locus elsewhere in the genome [59]. In contrast to viral gene addition, with insertion happening semi-randomly, the location of insertion can now be specifically chosen, eliminating the risk of insertional mutagenesis (Fig. 1).
In the absence of a homologous donor template, a cell can repair the DSB by nonhomologous end joining (NHEJ). NHEJ is error-prone and may therefore lead to allele inactivation, thus potentially correcting gain-of-function and dominant-negative mutations or inducing a therapeutic knockout [60]. NHEJ occurs at a much higher rate than homology directed repair. Therefore, allele inactivation is much easier to accomplish than correction with gene editing using engineered nucleases [61].
Even though engineered nucleases are designed to target a specific site, there's still the risk of introducing unwanted genomic alterations (on or off-site mutagenesis) leading to genotoxicity. Over the last decade new nuclease variants have been generated to reduce off- and on-target adverse effects [62,63] and tools to evaluate these risks [64]. Currently, homology directed repair is still inefficient and generally less efficient than viral gene addition, making it only suitable for diseases with a high selective advantage for corrected cells. In addition, delivering the (modified) nuclease and the donor template into the HSCs efficiently without losing the HSCs pluripotent potential is still challenging, especially when aiming to introduce an entire copy of a therapeutic gene and not just a short fragment to correct a specific region only. The last decade, several methods have been optimized to introduce the programmed nucleases efficiently to the cells [65], but the large size of the donor template makes most nonviral methods inadequate for the donor template. The most commonly used successful strategy has been delivery by AAV6 [45,47,48,49▪,51–53], an adeno-associated nonintegrating viral vector. The safety of this approach and the potential risk for AAV integration needs to be further evaluated before clinical translation [66▪].
Prime and base editing strategies are being developed that use impaired Cas or TALEN variants that edit the mutation directly, without introducing a DSB or nick and without the need for any donor template [67,68]. As a proof of concept, base editors have been used in patient cells to correct one of the most common heterozygous STAT3 mutations causing hyper IgE syndrome [69]. Although prime and base editors are appealing from a safety perspective, they require a different strategy for each mutation, making them less suitable as therapeutic option for most IEI as these are all rare diseases caused by many different mutations.
GENE THERAPY FOR SEVERE COMBINED IMMUNODEFICIENCYSevere combined immunodeficiencies (SCIDs) can be caused by different genetic defects resulting in defective differentiation of HSCs into T cells. Natural killer (NK) cells and B cells can be absent as well and although B cells can be present, those are always dysfunctional due to either intrinsic defects or lack of proper T cell help. These patients lack adaptive immunity completely and require curative treatment as early as possible or they will succumb to life-threatening infections within the first year of life. The implementation of TREC newborn screening in many countries has resulted in earlier detection and treatment [70].
Gene therapy is currently tested in clinical trials for ADA-SCID, X-linked SCID, Artemis SCID and RAG1 SCID. In Europe it is available as market authorized product for the treatment of ADA-SCID (Strimvelis).
Adenosine deaminase-deficient severe combined immunodeficiencyADA-SCID is characterized by absence of all lymphoid cells, due to enzyme deficiency and toxic accumulation. Additionally, skeletal, nervous system, and liver abnormalities are often observed in patients [71]. Treatment options for ADA-SCID patients include enzyme replacement therapy, HSCT and gene therapy [72]. Enzyme replacement therapy allows stabilization of patients with metabolic detoxification, but comes with less efficient long-term immune reconstitution and has high financial costs [72].
Since 2016, Strimvelis, a gRV vector based product, has been available as market approved gene therapy for ADA-deficient patients without HLA-matched donor [14]. In contrast to other SCID patients treated with first generation gRV vectors, the rate of subsequent cancer development is much lower in ADA-SCID. After three decades, only 1 of at least 50 patients with ADA-SCID treated with a gRV vector based gene therapy, either on clinical trial [9,25,26,73,74▪,75,76] or with Strimvelis, has developed leukemia [77]. Detailed analysis showed integration in or near proto-oncogenes in more patients, but this did not lead to any leukemia [75]. The mystery of why ADA-SCID patients appear to have a much lower risk of insertional oncogenesis despite the use of gRV vectors remains unsolved [78]. In parallel, more than 50 ADA-SCID patients have been treated successfully with an LV vector [79▪▪]. Recent studies comparing gene therapy to mismatched donor HSCT for ADA-SCID showed a favorable survival of 100% versus 79.6%, respectively, with GvHD occurring in 24% of the HSCT patients [74▪,76].
X-severe combined immunodeficiencyX-SCID is caused by pathogenic mutations in the X-linked gene IL2RG, that encodes the common gamma chain (γc) of the interleukin (IL)-2 receptor. Lack of γc-mediated signaling causes impaired development of T and NK cells, with presence of dysfunctional B cells. Initially, gRV vectors were used without conditioning, resulting in poor B cell reconstitution [7,12,13]. Six out of 20 treated patients developed acute T cell malignancies, due to integration near oncogenes, most notably LMO2 [12,34,36]. Further investigation showed that the therapeutic gene itself was not oncogenic [80]. This led to the development of safer SIN gRV viral vectors, with similar reconstitution as the previous gRV vectors [37]. Subsequent use of LV vector and low dose Busulfan conditioning led to improved T and B cell reconstitution [27,28]. Trials in the USA used an LV vector produced by St. Jude, with insulators flanking the codon optimized transgene. However, they showed clonal dominance in all 8 patients, which led to a voluntary hold of these trials (NCT01306019 and NCT01512888). Further safety analysis showed that the insulator used acts as a cryptic splice site resulting in truncated transcripts giving survival advantage to HMGA2 inserted clones [81]. Currently, two ongoing trials in the US and London are treating X-SCID patients with a different LV vector without any insulators in combinations with low dose busulfan conditioning (NCT03311503 and NCT03601286) with good B and T cell recovery [82,83].
Artemis-deficient severe combined immunodeficiencyArtemis-deficient SCID is an autosomal recessive form of T-B-NK+SCID caused by pathogenic mutations in the DCLRE1C gene, encoding Artemis. This nuclease is critical for nonhomologous end joining of gene segments from Ig and TCR genes during recombination. Artemis is required to complete the last step of V(D)J rearrangement of the B and T cell receptor and in general for repair of DNA double strand breaks. Therefore, in addition to susceptibility to severe infections, Artemis patients are more prone to the toxic effects of ionizing radiation and alkylating chemotherapy. In particular, Artemis patients seem to suffer from the long-term toxic effects of conditioning chemotherapy, yet reduced conditioning leads to poor B and T cell reconstitution [84,85]. This led to the development of gene therapy with LV. An initial trial using low dose Busulfan showed good reconstitution and no insertional mutagenesis during follow up of 8–48 months, but longer follow up will be essential [86▪▪].
Recombination activating genes 1/2 severe combined immunodeficiencyRecombination activating genes (RAG) 1 and 2 encode RAG1 and RAG2 proteins and are responsible for recombining the different V, (D) and J genes during B and T cell receptor rearrangement. Complete lack of functional RAG1 or RAG2 leads to T-B-NK+ SCID. As in other SCID genotypes, overall survival after HSCT with mismatched donors remains less favorable [17]. LV gene therapy with codon optimized RAG1 has been developed and after successful proof of concept results in RAG1 KO mice [87], a clinical trial has opened for RAG1-deficient SCID patients without an available HLA-matched donor (NCT04797260). The study is ongoing, with a favorable clinical course and immunological recovery in the first patients (unpublished results). For RAG2-SCID, a LV gene addition approach is being prepared for evaluation in a clinical trial (manuscript in preparation).
GENE THERAPY FOR OTHER IEI THAN SEVERE COMBINED IMMUNODEFICIENCY Wiskott–Aldrich syndromePathogenic mutations of the WAS gene cause an X-linked IEI resulting in defective actin polymerization and cytoskeletal remodeling affecting all hematopoietic lineages. This leads to complex immune dysregulation with eczema, thrombocytopenia and susceptibility to infections [88,89]. HSCT is the treatment of choice, with an overall survival of 89% [18]. The initial gene therapy trial for WAS used gRV vector and showed immunological reconstitution, but high incidence of insertional oncogenesis with seven out of nine patients developing leukemias, based on a similar mechanism as observed in the early X-SCID trials [32,33]. In subsequent studies with LV vectors no genotoxicity has been reported to date [90,91,92▪]. Immune improvement was favorable while platelet recovery was moderate, but high enough to prevent clinically significant bleeding.
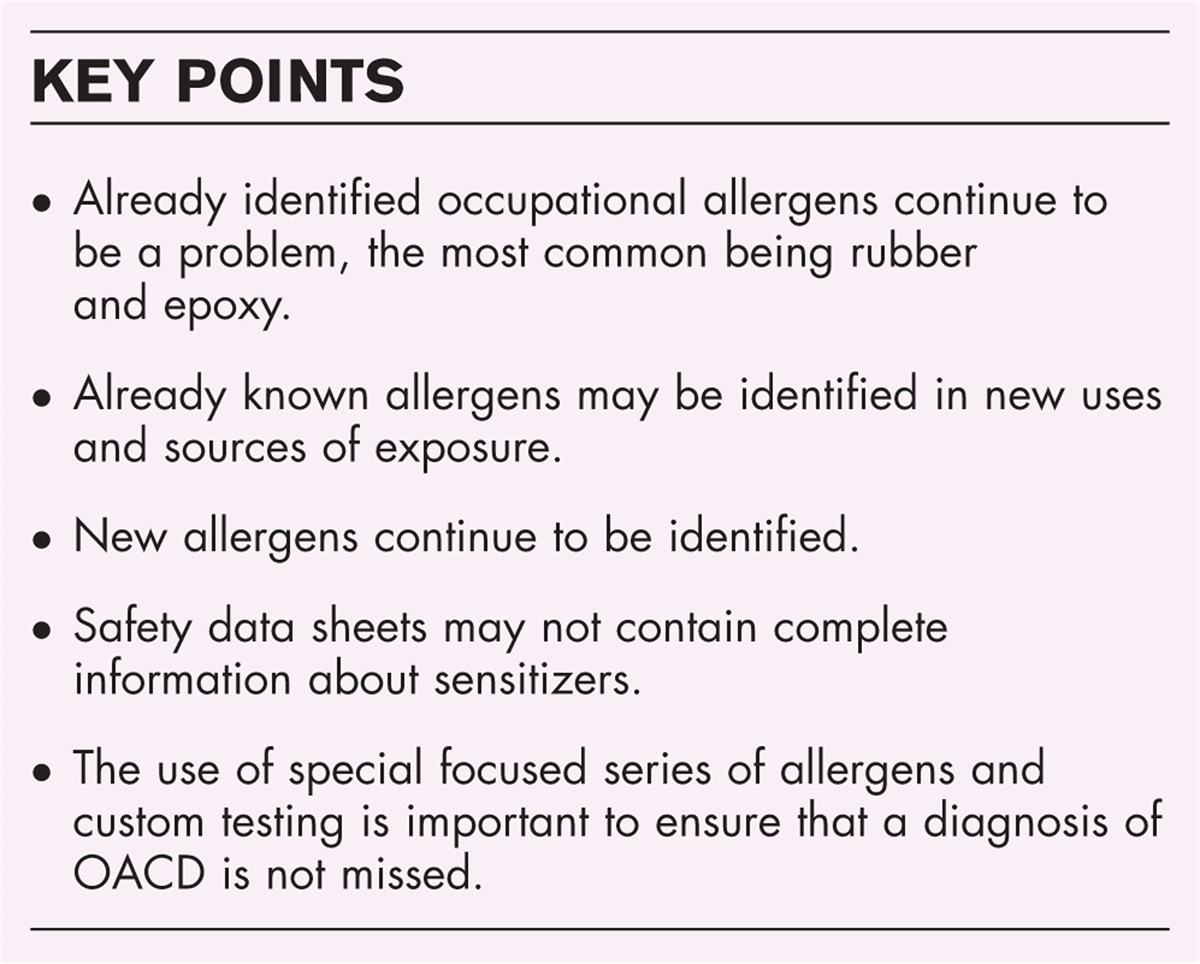
Neutrophil defectsChronic granulomatous disease (CGD) is caused by mutations in the genes encoding subunits of the nicotinamide adenine dinucleotide phosphate (NADPH) complex resulting in dysfunctional production of reactive oxygen species and defective phagocytic function. The most common form of CGD is X-linked CGD caused by mutations in the CYBB gene encoding the gp91phox component. Mutations in the genes encoding other subunits of the NADPH complex are inherited in an autosomal recessive manner, the most common form of autosomal recessive CGD is caused by mutations in NCF1. Patients with CGD present with life threatening bacterial and fungal infections as well as uncontrolled inflammatory complications, in particular colitis [93]. Treatment of CGD is based on lifelong antibacterial and antifungal prophylaxis, at times combined with IFN-gamma treatment [94]. HSCT is curative with an overall survival of 86% for patients younger than 18 years of age and favorable outcomes in patients receiving HLA-matched stem cells [21]. Initial gene therapy trials for X-CGD with gRV vectors that were conducted thirty years ago led to correction of neutrophil function but were also complicated by vector-induced myelodysplastic syndrome [95]. More recent trials used LV vectors and showed long-term functional correction in the majority of patients without any signs of genotoxicity [96]. However, all four surviving pediatric patients treated in this trial showed initial neutrophil recovery followed by declining levels of corrected cells over the next months [96]. Another trial using LV vector showed a similar phenomenon and additional studies suggest that the inflammation affects quality and engraftment potential of the HSCs [97,98▪▪]. For autosomal recessive CGD (p47phox), preclinical studies in mice showed effective use of LV vector and a clinical trial has recently opened at the Great Ormond Street Hospital for Children with plans for an additional trial at the National Institutes of Health (NIH) (Table 1).
Leukocyte adhesion deficiency type 1 (LAD-1) is caused by recessive pathogenic mutations in the ITGB2 gene, resulting in impaired expression of CD18, a crucial integrin molecule for neutrophil adhesion and migration [99]. Untreated patients develop early and severe bacterial infections with no purulent exudate formation as well as long term severe autoimmune complications [100]. HSCT leads to 75% overall survival [101]. Preclinical studies in animal models showed feasibility of gene therapy with a viral vector [102,103]. However, initial trials using gRV vector were unsuccessful and terminated early as no corrected cells were detected [104]. Currently, trials are being conducted using LV vectors in combination with busulfan conditioning, with interim reports showing successful reversal of the LAD-1 phenotype [105,106].
Immune dysregulation, polyendocrinopathy enteropathy X-linked syndromeImmune dysregulation, polyendocrinopathy enteropathy X-linked syndrome (IPEX) is caused by pathogenic mutations in the FOXP3 gene. FOXP3 is a transcription factor required for the development of regulatory T cells (Tregs). Consequently, the syndrome is characterized by enteropathy and multiple autoimmune features. Treatment consists of supportive care, immune suppression and HSCT [107,108]. Using an LV vector to express FOXP3 in HSCs appeared to prevent T cells from differentiating into all required peripheral subsets [109]. However, mouse studies showed that using an LV-FOXP3 vector in CD4+ T cells resulted in functional Tregs and rescue of the scurfy phenotype [110,111]. Based on these preliminary studies, there is an open clinical trial for patients with IPEX syndrome at Lucile Packard Children's Hospital Stanford (Table 1).
Hypomorphic recombination defectsWhen mutations in DCLRE1C or RAG1 and 2 lead to residual activity, they can cause a broad immunological and clinical spectrum with different degrees of immunodeficiency and autoimmunity or inflammation [112–115]. HSCT is potentially curative in these patients, but more challenging than in individuals with SCID, because of increased risk for graft rejection due to residual T cell function. In addition, thymic abnormalities affecting mechanisms of immune tolerance have been described in patients with hypomorphic RAG mutations [116–118]. A recent retrospective study of HSCT in patients with hypomorphic RAG defects showed that poor pre-HSCT clinical status predicted an unfavorable HSCT outcome and slower naïve T-cell recovery, with especially low overall survival in the absence of an HLA matched donor [20]. Gene therapy might provide a strategy to overcome these hurdles. However, in patients with hypomorphic RAG deficiency uncorrected HSCs may retain the capacity to develop into defective peripheral T and B cells, a problem that is not encountered in uncorrected HSCs from SCID patients where there is a complete developmental block at an early progenitor stage [115].
CHALLENGES AND FUTURE PERSPECTIVESFrom 1995 to 2020 more than 200 patients with IEIs have been treated with gene therapy using viral gene addition, initially with gRV, later with self-inactivating (SIN) variants of gRV and LV [119]. These newer SIN vectors have been shown to be safer, with no hematological malignancies reported as yet in IEI patients. With improved protocols on HSC manipulation and preparative conditioning improving immune reconstitution, the number of successful gene therapy trials in IEI has grown significantly over the last decade. However, several challenges remain. Genes that are tightly regulated can be harmful when constitutively expressed with viral gene addition, as was shown for CD40L [57]. In such cases, gene editing with engineered nucleases would offer a promising alternative as it preserves endogenous gene regulation. Unfortunately, gene editing approaches tailor made for correction of a specific mutation are inefficient for IEI as patients generally show unique mutations and would each require a different gene therapy product. To date, several research groups have shown proof of principle for gene editing to correct IEI by adding an entire copy of the therapeutic gene to the same locus under the control of its endogenous promoter. In that case, the same approach would be applicable to all mutations. However, in contrast to delivery of the engineered nuclease, delivery of donor template is still challenging and has mostly been accomplished by use of nonintegrating viral vectors (AAV6) requiring additional safety studies [45,47,48,49▪,51–53,66▪].
For IEI that mainly affect T cells, modifying T cells is a good alternative. For IPEX, viral gene addition is being tested in a clinical trial [110,111] (Table 1). Gene editing of T cells instead of HSCs could be an attractive alternative as well. Gene editing of T cells reduces the risk of deleterious off-target mutagenesis as the cells are terminally differentiated and require less toxic conditioning for engraftment [120]. Proof of concept has been shown for CD40L deficiency [49▪], CTLA4 [52], IPEX [121] and XLP [53]. These studies also used donor template delivery by AAV6.
When gene therapy has been proven safe and effective in clinical trials, the next step is to make the therapy more widely available. Except for hospital exemption, currently the only option is to get market authorization and subsequent reimbursement. Gene therapy for ADA-SCID, Strimvelis (Orchard Therapeutics), was the first (and so far only) therapy to get this authorization in the EU. Remarkably, it was not reimbursed in all EU countries. Last year Orchard Therapeutics announced that they will discontinue gene therapy for WAS, X-CGD and ADA-SCID for commercial reasons [122▪▪,123]. It is becoming increasingly clear that the current for-profit model of market authorization and commercialization is not economically fit to achieve sustainable and affordable access to gene therapy for rare diseases such as IEI. To find solutions to these challenges the AGORA (Access to Gene Therapies for Rare Diseases) initiative was started comprising clinical academics, scientists and patient organizations [124] and a number of other national and international efforts are trying to obtain the same goal.
CONCLUSIONA series of gene therapy trials using viral gene addition have been successful and gene therapy for ADA-SCID has been market authorized in Europe. Viral gene therapy approaches have become safer, more efficient and applicable to more IEI. Still, major obstacles for their further implementation and spread to many countries are health economics and complex regulations. Gene editing strategies to treat IEI are still at a preclinical level, but are promising alternatives for tightly regulated genes, gain-of-function mutations or dominant-negative mutations. Challenges for gene editing are minimizing on and off-site mutagenesis and optimizing correction efficiency. Current gene editing correction strategies applicable to multiple different mutations require a donor template containing the therapeutic gene. Delivering the donor template safe and efficiently to HSCs is still challenging.
AcknowledgementsNone.
Financial support and sponsorshipOur lab work is supported in part by a ZonMW E-RARE grant (40-419000-98-020) and EU H2020 grant RECOMB (755170-2) and has received funding from the European Union Horizon 2020 research and innovation program as well as from reNEW, the Novo Nordisk Foundation for Stem Cell Research (NNF21CC0073729).
Conflicts of interestL.O.B. receives grant support from NovoNordisk. FJTS receives grant support from Batavia Biosciences, NovoNordisk and Mustang Bio in accordance with the rules and guidelines by the Dutch Federations of Universities (NFU).
REFERENCES AND RECOMMENDED READINGPapers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES 1. Tangye SG, Al-Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 2022; 42:1473–1507. 2. Gatti RA, Meuwissen HJ, Allen HD, et al. Immunological reconstitution of sex-linked lymphopenic immunological deficiency. Lancet 1968; 2:1366–1369. 3. De Koning J, Van Bekkum DW, Dicke KA, et al. Transplantation of bone-marrow cells and fetal thymus in an infant with lymphopenic immunological deficiency. Lancet 1969; 1:1223–1227. 4. Hirschhorn R, Yang DR, Puck JM, et al. Spontaneous in vivo reversion to normal of an inherited mutation in a patient with adenosine deaminase deficiency. Nat Genet 1996; 13:290–295. 5. Stephan V, Wahn V, Le Deist F, et al. Atypical X-linked severe combined immunodeficiency due to possible spontaneous reversion of the genetic defect in T cells. N Engl J Med 1996; 335:1563–1567. 6. Staal FJT, Aiuti A, Cavazzana M. Autologous stem-cell-based gene therapy for inherited disorders: state of the art and perspectives. Front Pediatr 2019; 7:443. 7. Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 2000; 288:669–672. 8. Aiuti A, Slavin S, Aker M, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science 2002; 296:2410–2413. 9. Aiuti A, Cattaneo F, Galimberti S, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med 2009; 360:447–458. 10. Blaese RM, Culver KW, Chang L, et al. Treatment of severe combined immunodeficiency disease (SCID) due to adenosine deaminase deficiency with CD34+ selected autologous peripheral blood cells transduced with a human ADA gene. Amendment to clinical research project, Project 90-C-195, January 10, 1992. Hum Gene Ther 1993; 4:521–527. 11. Kohn DB, Weinberg KI, Nolta JA, et al. Engraftment of gene-modified umbilical cord blood cells in neonates with adenosine deaminase deficiency. Nat Med 1995; 1:1017–1023. 12. Hacein-Bey-Abina S, Le Deist F, Carlier F, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med 2002; 346:1185–1193. 13. Gaspar HB, Parsley KL, Howe S, et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet 2004; 364:2181–2187. 14. Aiuti A, Roncarolo MG, Naldini L. Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: paving the road for the next generation of advanced therapy medicinal products. EMBO Mol Med 2017; 9:737–740. 15. Pai SY, Logan BR, Griffith LM, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med 2014; 371:434–446. 16. Gennery AR, Lankester A, Marrow T. Inborn Errors Working Party of the European Society for B. Long term outcome and immune function after hematopoietic stem cell transplantation for primary immunodeficiency. Front Pediatr 2019; 7:381. 17. Lankester AC, Neven B, Mahlaoui N, et al. Hematopoietic cell transplantation in severe combined immunodeficiency: the SCETIDE 2006–2014 European cohort. J Allergy Clin Immunol 2022; 149:1744–1754. e8. 18. Albert MH, Slatter MA, Gennery AR, et al. Hematopoietic stem cell transplantation for Wiskott–Aldrich syndrome: an EBMT Inborn Errors Working Party analysis. Blood 2022; 139:2066–2079. 19. Dedieu C, Albert MH, Mahlaoui N, et al. Outcome of chronic granulomatous disease – conventional treatment vs stem cell transplantation. Pediatr Allergy Immunol 2021; 32:576–585. 20. Schuetz C, Gerke J, Ege M, et al. Hypomorphic RAG deficiency: impact of disease burden on survival and thymic recovery argues for early diagnosis and HSCT. Blood 2023; 141:713–724. 21. Chiesa R, Wang J, Blok HJ, et al. Hematopoietic cell transplantation in chronic granulomatous disease: a study of 712 children and adults. Blood 2020; 136:1201–1211. 22. Tajer P, Pike-Overzet K, Arias S, et al. Ex vivo expansion of hematopoietic stem cells for therapeutic purposes: lessons from development and the niche. Cells 2019; 8:
Comments (0)