
Remember me



The patient (Fig. 1, IV-1) was a 13-year-old boy with unaffected first-cousin parents. At birth, he had normal growth parameters. However, he began to suffer from feeding difficulties, respiratory distress, brain anomalies, and hypotonia, and consequently was admitted to neonatal intensive care unit for two weeks. Physical examination revealed developmental delay (including jerking movements of the arms and legs that cannot be controlled), distinctive facial features, epilepsy and tonic seizures, chest infections, and mental abnormalities. Phenotypes of the patient included thick eyebrows, hypertelorism, short nose, upslanted palpebral fissures, thick lips, broad nasal bridge, upturned nasal tip, broad and low-hanging columella, low-set ears, high-arched palate, coarse facies, and flat occiput. His developmental delay consisted of slower-than-normal development of motor, cognitive, social, and emotional skills. Additionally, the child exhibited pronounced hypotonia, often described as a 'strong floppy' condition, and the recurrent chest infections were attributed to respiratory issues.
Brain MRI showed white matter atrophy in parieto-occipital lobes with abnormal periventricular T2 hyperintensity returned from the deep white matter, and foreshortening and thinning of a corpus callosum (Fig. 2).
Fig. 2
Brain MRI of the patient (right to left sagittal T1, axial T2 and coronal T2 images) showing mild ventricular hypoplasia with prominent CSF spaces, reduced white matter volume, hypoplasia of corpus callosum, and cavum septum pellucidum
Interestingly, his deceased sister (Individual IV-2 in the pedigree, Fig. 1) was reported to have the same clinical symptoms and phenotypes. Her clinical course included the presence of muscle weakness, seizures, hypotonia, feeding difficulties, respiratory distress, developmental delay, and mental retardation. Unfortunately, no DNA sample was available from her and no molecular examination was possible.
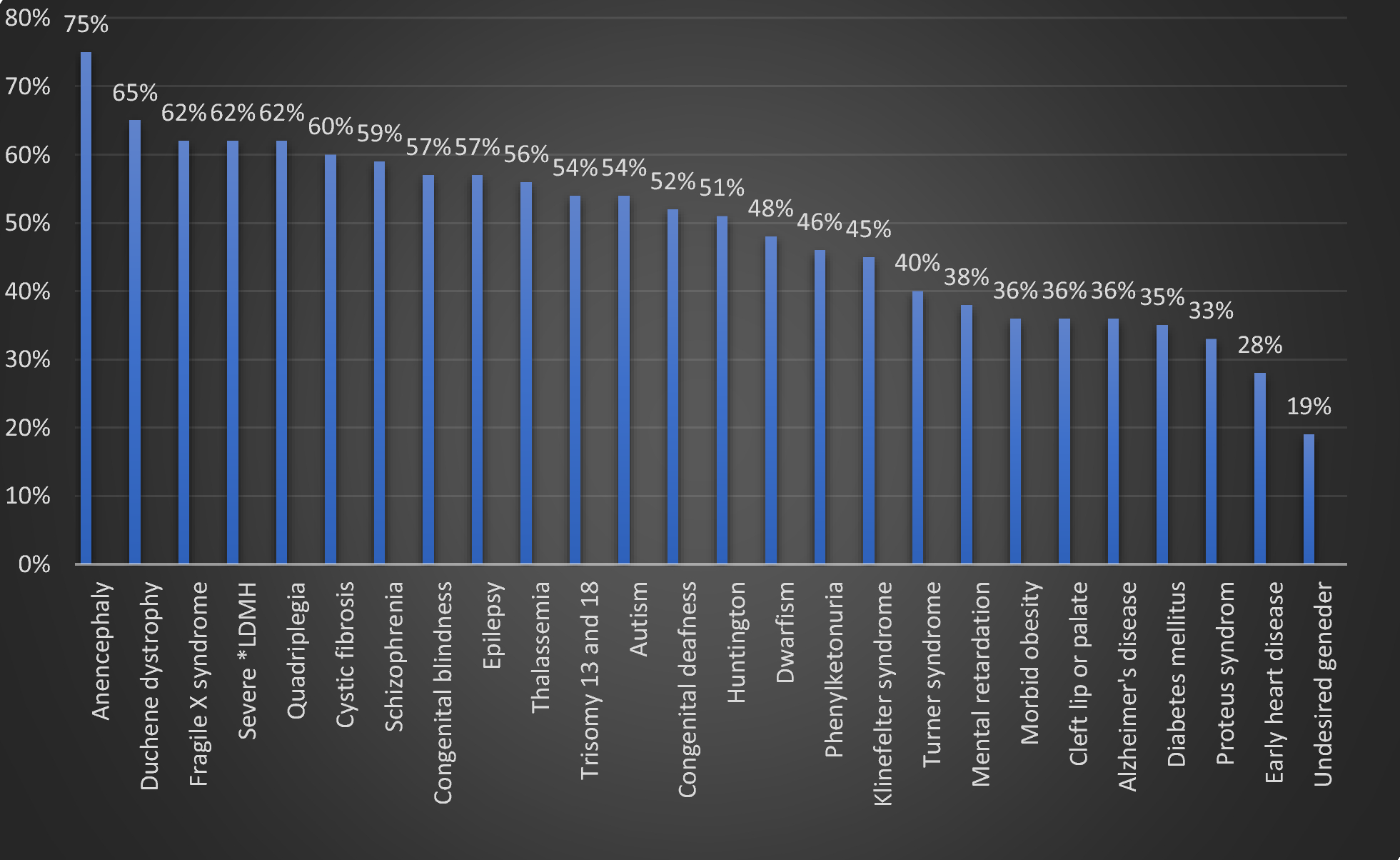
Genetic findings and bioinformatics analysisWES was performed on the patient (Fig. 1, IV-1). After quality control and alignment steps, a number of 72,695 variants were called by GATK4. Subsequently, annotation of these variants was performed with ANNOVAR. In particular, the homozygous alterations (21,815 variants) were considered because of apparently recessive mode of inheritance. In the filtering process, by excluding the variants with allele frequency of greater than 1%, only 2071 homozygous variants remained. Then, synonymous and benign variants were excluded, which ended up with 314 variants. Next, we chose only variants located in the patient’s phenotype-related genes (19 variants). The details of these filtering steps are shown in Fig. 3.
Fig. 3
Flowchart of variant filtering process performed through whole exome sequencing (WES) data analysis. After annotation, variants were filtered based on described parameters, and finally one disease-causing variant in PPP1R21 gene was found
Variants were interpreted based on clinical significances and in-silico predictions. We detected one rare homozygous frameshift variant (Fig. 4), which was a deletion of two nucleotides, c.1317_1318delAG; p.(Asp440Tyrfs*6) in exon 13 of PPP1R21 gene (NM_001135629.3). According to the American College of Medical Genetics (ACMG) guidelines [15], this loss-of-function variant is classified as a pathogenic mutation. The ACMG evidences in support of its pathogenicity are as follows:
Fig. 4
A schematic section of the PPP1R21 gene, and the location of the identified pathogenic variant (c.1317_1318delAG) in human genome (GRCh38/hg38 assembly). Conservation of the region containing the mutation across different species is shown as well
PVS1 This mutation is a null (frameshift) variant in PPP1R21 gene, for which loss-of-function is a known mechanism of disease. The identified variant p.(Asp440Tyrfs*6) would lead to a change in the protein structure by disturbing normal splicing and creating a stop codon at amino acid (AA) 445 instead of AA 781 in wild type sequence, causing nonsense-mediated mRNA decay and complete loss of function;
PM2 This PPP1R21 variant has not been yet described and was not found in any population database including gnomAD, dbSNP, ExAC, and Iranome databases;
PS4 The variant was seen in affected family members and was not detected in the normal controls;
PP4 Patient's phenotype and family history is highly specific for a neurodevelopmental disease with a single genetic etiology;
PM4 This variant reduces the length of the protein;
PP1 The variant is segregated with the disease in affected family members; and
PP3 In silico prediction algorithms, including SIFT, PolyPhen2, MutationTaster, Franklin, and VarSome, strongly support the pathogenic and deleterious effects of this mutation.
Sanger sequencing confirmed the homozygous status in the patient and the heterozygous status in his parents (Fig. 5). Thus, the parents were obligate carriers. In conclusion, results showed the co-segregation of the c.1317_1318delAG variant in PPP1R21 gene with the disease in the family.
Fig. 5
Sequencing chromatogram of genomic DNA showing c.1317_1318delAG; p.(Asp440Tyrfs*6) mutation in PPP1R21 gene. The patient was homozygous for this variant, and the parents were heterozygous
The protein cluster network of STRING database for PPP1R21 protein was performed, and it showed the highest predicted association scores with C2orf44, PIBF1, TMEM247, CRYZL1, STON1, PCDHGA10, and STRN genes. Likewise, multispecies alignment for this variant indicated high conservation within species (Fig. 6).
Fig. 6
The in-silico analysis of the functional protein association network was assessed via online STRING server for PPP1R21 interactions
Comments (0)