
Remember me
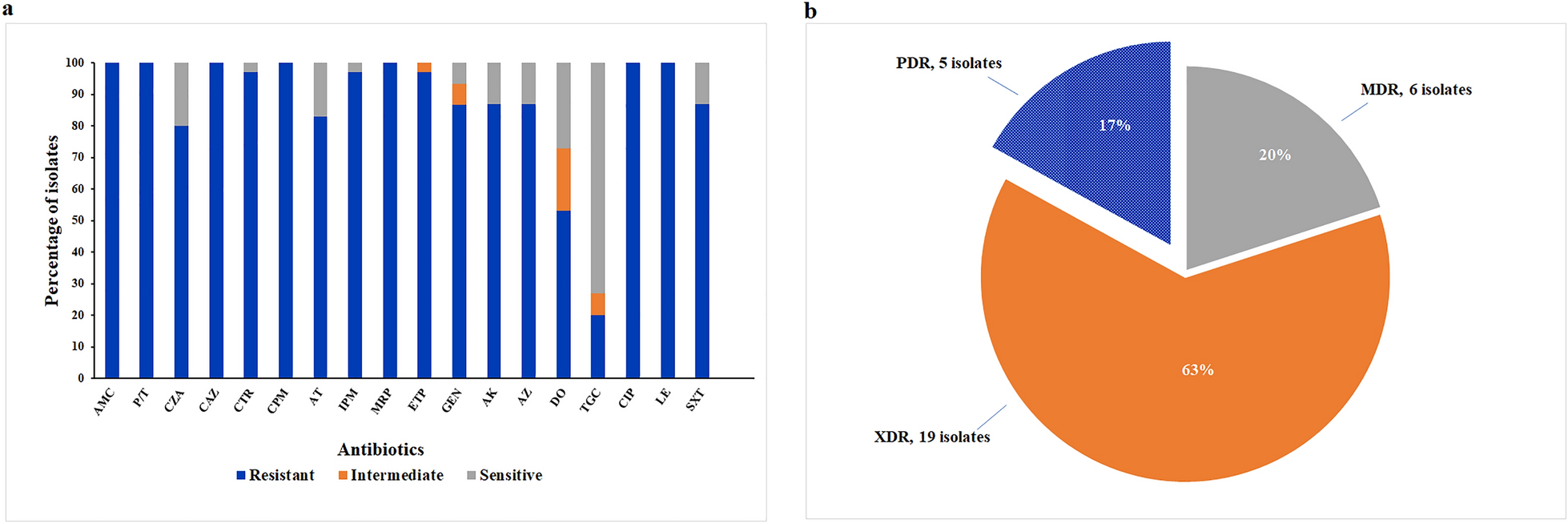
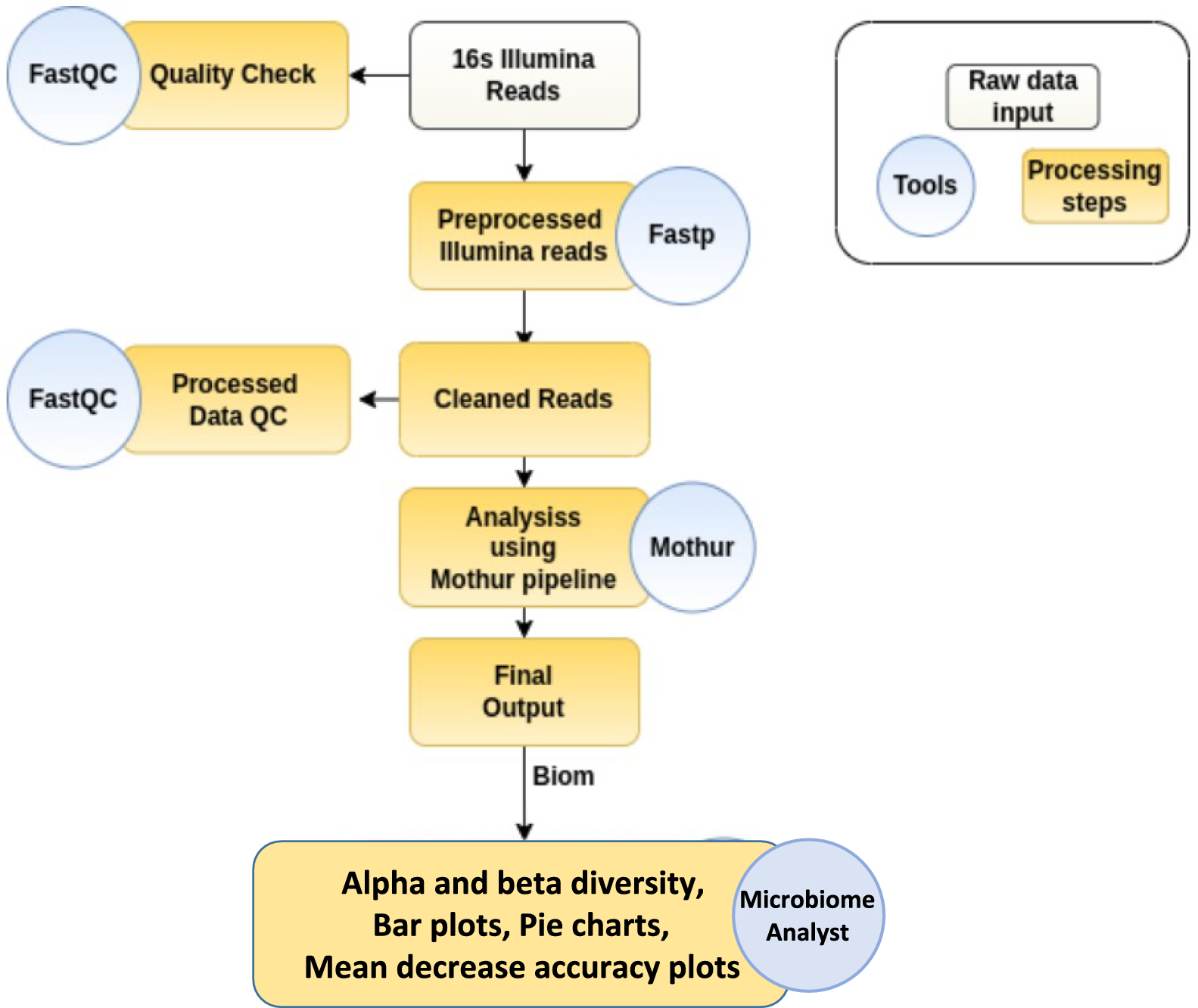

A total of 105 Fusobacterium nucleatums’ genome sequences were downloaded from the NCBI, quality controlled and evaluated for core genes. Finally, 93 genomes were selected for further analysis (Additional file 5: Table S1). The number of scaffolds ranged from 1 to 379, with a maximum N50 value of 2,653,055 bp, a minimum value of 8,680 bp and an average genome size of 2,369,555 bp. We collected the metadata of the downloaded strains (Additional file 6: Table S2) and found that Fusobacterium nucleatum was isolated mainly from the oral cavity (N = 33) and the intestine (N = 16), excluding the majority of strains with unknown phenotypes.
Pan-genomic characterization of Fusobacterium nucleatumA total of 93 Fusobacterium nucleatum strains were included in the first pangenome analysis. The Fusobacterium nucleatum genome contains a total of 21,139 gene families, of which 516 are core (present in more than 95% of the genome). The number of variable gene families is 20,623. According to Fig. 1, the pan-genome shows a clear open tendency, and the size of the pan-genome continues to increase with the number of genomes included in the analysis, showing a continuous upward trend. The number of emerging gene families in the pan-genome increases with the number of genomes, and in turn the size of the pan-genome will expand. The heatmap of gene presence-absence matrix showed two distinct clades in Fusobacterium nucleatum (Additional file 1: Figure S1).
Fig. 1
The pan genome plot of Fusobacterium nucleatum. A. Conserved genes and Total genes. B. New genes and Unique genes
Based on the core gene set, we constructed a cgmlst molecular marker (N = 298) for Fusobacterium nucleatum (Additional file 7: Table S3A) and a phylogenetic tree for 93 strains based on this markers (Additional file 2: Figure S2). The phylogenetic tree showed that there were no obvious clades of Fusobacterium nucleatum and, based on the known meta information, the strains from the oral cavity as well as the intestine were scattered and did not aggregate significantly. Functional enrichment of these 298 genes showed that they were mainly derived from the Ribosome and ABC transporters pathways (Additional file 3: Figure S3). Notably, we also attempted to construct separate cgmlst molecular markers from the oral cavity and intestine (Additional file 7: Table S3BC), and the Venn diagram shows that these two types of markers share 384 genes, while the oral cavity (N = 16) and intestine (N = 161) each retain a small number of cgmlst genes (Additional file 4: Figure S4).
Bioinformatic analysis of virulence genes and FadA geneWe examined the virulence genes in the genomic data of 93 Fusobacterium nucleatum strains based on the VFDB database (Fig. 2). A total of 11 virulence genes were found to be present in the genome, notably groEL, clpP and acpXL were found to be present in 93 strains with copy number 1. tufA was present in most strains, while other virulence factors such as cap8E, neuB and wbtE were present in a small number of strains. In addition, we also predicted drug-resistant genes for these strains and found that the majority of Fusobacterium nucleatum did not carry those genes, but were present in only a few strains.
Fig. 2
Heatmap of virulence related genes in Fusobacterium nucleatum
We also analyzed the Fusobacterium nucleatum genomes for the FadA genes, a hair adhesion protein that is important for cell binding. We found that 90 of these strains contained the FadA gene in their genome sequences and, based on the FadA protein sequence, we constructed a phylogenetic tree that showed three distinct clades of the FadA gene, with strains from the oral and intestinal tracts in each of the three clades (Fig. 3A). In addition, we investigated the upstream and downstream structure of the FadA gene and found that the upstream and downstream structure of the FadA gene is relatively conserved in Fusobacterium nucleatum genomes, with the FadA gene surrounded by ABC transporter permease and Peptidylprolyl isomerase, and upstream and downstream genes such as EnvC and NAD kinase (Fig. 3B).
Fig. 3
Genomic analysis of FadA genes in Fusobacterium nucleatum. A. The phylogenetic tree of FadA genes. B. the genomic structure of FadA genes
Plasmid prediction and genomic analysis of Fusobacterium nucleatumWe have used the newly developed plasmid prediction tool Plasmer to predict the genome sequences of Fusobacterium nucleatum. In total, we found plasmid sequences in the genomes of 42 strains. We then filtered plasmid sequences with high quality genomes for subsequent analysis (number of contigs < 3) and validated the plasmids based on the NCBI non-redundant nucleic acid library. In total, we identified 17 strains with relatively complete plasmid sequences present. Of these plasmid genomes, 13 are known, and in addition we identified four unreported sequences of around 15 K in length, which we speculate are likely to be newly discovered plasmid sequences (Table 1). Among the known plasmid genomes, five strains carry plasmid type 7–1, while other plasmid types include 4–8, pFN3 and pCT15E1. 7–1 plasmid has a genome size of 6.3 K and contains a total of seven mRNA-encoding genes, most of which are putative proteins, with no resistance or virulence genes identified (Fig. 4).
Table 1 The predicted plasmids of Fusobacterium nucleatumFig. 4
The circular representation map of 7–1 plasmid
Comments (0)