
Remember me
Pancreatic cancer is the third most common cause of cancer-related deaths in the United States. Although gemcitabine is the standard of care for most patients with pancreatic cancer, its efficacy is limited by the development of resistance. This resistance may be attributable to the evasion of apoptosis caused by the overexpression of BCL-2 family antiapoptotic proteins. In this study, we investigated the role of BCL-XL in gemcitabine resistance to identify a combination therapy to more effectively treat pancreatic cancer. We used CRISPR-Cas9 screening to identify the key genes involved in gemcitabine resistance in pancreatic cancer. Pancreatic cancer cell dependencies on different BCL-2 family proteins and the efficacy of the combination of gemcitabine and DT2216 (a BCL-XL proteolysis targeting chimera or PROTAC) were determined by MTS, Annexin-V/PI, colony formation, and 3D tumor spheroid assays. The therapeutic efficacy of the combination was investigated in several patient-derived xenograft (PDX) mouse models of pancreatic cancer. We identified BCL-XL as a key mediator of gemcitabine resistance. The combination of gemcitabine and DT2216 synergistically induced cell death in multiple pancreatic cancer cell lines in vitro. In vivo, the combination significantly inhibited tumor growth and prolonged the survival of tumor-bearing mice compared with the individual agents in pancreatic cancer PDX models. Their synergistic antitumor activity is attributable to DT2216-induced degradation of BCL-XL and concomitant suppression of MCL-1 by gemcitabine. Our results suggest that DT2216-mediated BCL-XL degradation augments the antitumor activity of gemcitabine and their combination could be more effective for pancreatic cancer treatment.
IntroductionPancreatic cancer is one of the most aggressive human cancers with a 5-year survival rate of approximately 9% and a median survival of <11 months in the United States (1–3). It is the third most common cause of cancer-related deaths. Gemcitabine, a deoxycytidine analogue that inhibits DNA replication, is currently the first-line standard of care chemotherapy for pancreatic cancer. Unfortunately, the therapeutic efficacy of gemcitabine is limited by the innate and acquired resistance leading to treatment failure and recurrent disease in most patients (4–7). Although FOLFIRINOX (a combination of 5-fluorouracil, leucovorin, irinotecan, and oxaliplatin) treatment has increased the survival of patients with high-grade pancreatic cancer compared with gemcitabine, it is associated with increased toxicities and a decreased quality of life (8, 9). Therefore, the identification of the molecular basis of gemcitabine resistance and developing rational combination therapies that can more effectively treat pancreatic cancer are of a critically unmet medical need.
BCL-2 family antiapoptotic proteins (BCL-2, BCL-XL, and MCL-1) are involved in cancer progression and resistance to chemotherapy and radiation. Among these antiapoptotic proteins, BCL-XL plays a crucial role in pancreatic cancer (5, 10, 11). Previous reports have shown that in pancreas-specific KRASG12D mice, BCL-XL expression was gradually increased during the progression of pancreatic intraepithelial neoplasia (PanIN) to pancreatic ductal adenocarcinoma (PDAC; ref. 12). Another study showed that tumor tissues from approximately 90% of patients with pancreatic cancer expressed increased levels of BCL-XL (13). These high levels of BCL-XL in pancreatic cancer have been shown to be associated with gemcitabine resistance; therefore, BCL-XL inhibition sensitizes pancreatic cancer cells to gemcitabine treatment (14–17). Unfortunately, the direct targeting of BCL-XL with a conventional small-molecule inhibitor is not a clinically viable approach due to the on-target and dose-limiting thrombocytopenia caused by BCL-XL inhibition in platelets (18, 19). Recently, using emerging Proteolysis Targeting Chimera (PROTAC) technology, we successfully converted ABT263 (a BCL-XL/2 inhibitor) into DT2216, a platelet-sparing BCL-XL-selective PROTAC that targets BCL-XL to the Von Hippel-Lindau (VHL) E3 ligase for ubiquitination and proteasomal degradation (20). DT2216 has shown promising antitumor activities in BCL-XL–dependent hematologic malignancies when used as single-agent therapy and in multiple solid tumors when combined with conventional chemotherapy (20–22). Therefore, it has recently been authorized as an investigational new drug for dose-finding clinical trials by the FDA (NCT04886622). The lack of single-agent efficacy of DT2216 in solid tumors is mainly due to redundant expression of BCL-2, BCL-XL, and MCL-1 in these tumors, which makes them codependent on two or more of these proteins (23–26). This is because the interaction of proapoptotic proteins (e.g., BIM) with multiple partners may lead to these proapoptotic proteins switching interaction between different antiapoptotic proteins upon single inhibitor treatment. For instance, the displacement of BIM from BCL-XL and BCL-2 by ABT263 treatment led to its increased association with MCL-1 (27, 28). These findings underscore the importance of cotargeting multiple antiapoptotic BCL-2 proteins for effective antitumor therapy.
To identify druggable genes important to and commonly involved in the development of gemcitabine, 5-fluorouracil (5FU), and niraparib resistance in pancreatic cell lines, we conducted CRISPR screening using a custom library targeting drug targets, termed the Therapeutic Genome (RxG) Library. We found the BCL2L1 gene that encodes BCL-XL is important in providing resistance to these three agents. We selected gemcitabine to study in combination with DT2216 against various established and primary pancreatic cancer cells in vitro and in vivo in a xenograft mouse model, as well as in patient-derived xenograft (PDX) mouse models. We observed that the DT2216– gemcitabine combination synergistically kills various pancreatic cancer cells in vitro. More importantly, the combination was found to be more effective than the individual agents at inhibiting tumor growth and increasing the survival of the mice without causing any significant normal tissue toxicities. Our mechanistic investigation revealed that the downregulation of MCL-1 by gemcitabine coupled with BCL-XL degradation by DT2216 are implicated in their synergistic activity. Collectively, our results suggest that the combination of gemcitabine and DT2216 can synergistically suppress pancreatic tumor growth, and therefore, may reduce resistance to gemcitabine.
Materials and MethodsCell cultureAsPC-1 (catalog No. CRL-1682), BxPC3 (catalog No. CRL-1687), Mia PaCa2 (catalog No. CRL-1420), and PANC-1 (catalog No. CRL-1469) human pancreatic cancer cell lines were purchased from the ATCC. G-46 and G-68 patient-derived primary pancreatic cancer cell lines were established, characterized, and authenticated using methods described previously (29, 30). PANC-1 and Mia PaCa2 cells were cultured in DMEM (catalog No. 12430054, Thermo Fisher Scientific). AsPC-1 and BxPC3 cells were cultured in RPMI medium (catalog No. 22400–089, Thermo Fisher Scientific). G-46 and G-68 cells were cultured in advanced DMEM medium (catalog No. 12430054, Thermo Fisher Scientific). All the cell lines were cryopreserved from early passages and were cultured for no more than 12 passages following thawing. Cell lines were authenticated prior to use by short tandem repeat (STR) profiling. Cultures were confirmed for Mycoplasma negativity using the MycoAlert Mycoplasma Detection Kit (catalog No. LT07–318, Lonza). All culture mediums were supplemented with 10% heat-inactivated FBS (catalog No. S11150H, Atlanta Biologicals), 100 U/mL penicillin and 100 μg/mL streptomycin (Pen-Strep, catalog No. 15140122, Thermo Fisher Scientific). All the cell lines were maintained in a humidified incubator at 37° C and 5% CO2.
Chemical compoundsDT2216 was synthesized in Dr. Guangrong Zheng's laboratory (University of Florida, Gainesville, FL) according to the previously described protocol (20). Gemcitabine (catalog No. S1714), 5FU (catalog No. S1209), niraparib (catalog No. S2741), A1155463 (catalog No. S7800), ABT199 (catalog No. S8048), S63845 (catalog No. S8383), and ABT263 (catalog No. S1001) were purchased from SelleckChem. All the compounds were dissolved in DMSO at 10 mmol/L stock solution for in vitro assays.
CRISPR screening using the therapeutic genome (RxG) libraryBriefly, we designed a custom sgRNA library, the RxG library, targeting 996 genes for which therapeutic interventions have been identified (Supplementary Methods and Supplementary Excel File). We carried out CRISPR screens with the RxG library in the AsPC-1 cell line to identify candidate genes, which when targeted for disruption modulate sensitivity to three chemotherapeutics, gemcitabine, 5FU, and niraparib. Each screen was carried out in triplicate (IC30-IC50 of each drug) for 16 days (T16D) at approximately 500X library coverage. Triplicate puromycin selected samples (T0 PURO) prior to exposure without treatment were collected as controls. Cells from each screen were harvested, genomic DNA prepared, sgRNAs amplified, and quantified by next-generation sequencing (NGS). MAGeCK, which utilizes a-RRA (Robust Ranking Algorithm) to rank candidate genes, was used to identify candidate genes for which the corresponding sgRNAs showed significantly altered distribution between treated and T0 PURO control samples (31). Additional details are provided in the Supplementary Methods, and the CRISPR screening raw data are accessible at Dryad (https://doi.org/10.5061/dryad.m905qfv22).
Animal studiesNOD-scid IL2Rgammanull (NSG) mice aged 5 to 6 weeks were purchased from The Jackson Laboratory (Stock No. 005557). The mice were allowed to acclimatize for 1 week. Animals were housed in the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited animal facilities at the University of Florida under pathogen-free conditions. All animals received food and water ad libitum. G-68 PDX cells at 5 × 106 per mouse in 50% Matrigel (catalog No. 356237, Corning) in PBS were injected subcutaneously (s.c.) into the right flank region of the NSG mice (30). Pancreatic cancer PDX models (G192-p4, G176-p4, and LM12-p3) were established and characterized by Dr. Trevino at the University of Florida, and were propagated in NSG mice as reported previously (32). The tumors were harvested after they reached 1,500 mm3, cut into 2-mm fragments, and were implanted subcutaneously after submerging in Matrigel in additional NSG mice (32). Tumor size was measured twice a week with digital calipers, and tumor volume was calculated using the formula (length×width2×0.5) as described previously (20). The animals were randomized into different treatment groups when the tumors reached 100 to 200 mm3. Animals were treated with vehicle, gemcitabine [20 mg/kg, once a week (every 7 days), i.p.], DT2216 [15 mg/kg, every 4 days, i.p.] and a combination of gemcitabine and DT2216. Gemcitabine was dissolved in normal saline, and DT2216 was formulated in 50% phosal 50 PG, 45% miglyol 810N and 5% polysorbate 80. They were administered in 100 μL of vehicle (saline for gemcitabine and 50% phosal 50 PG, 45% miglyol 810N, and 5% polysorbate 80 for DT2216) per mouse. DT2216 treatment was initiated 2 days before gemcitabine treatment and continued as described previously. Mice were euthanized when they became moribund, or their tumor sizes reached humane endpoint as per Institutional Animal Care and Use Committee (IACUC) policy. For euthanasia, animals were sacrificed by CO2 suffocation followed by cervical dislocation. The tumors were subsequently harvested, fixed in buffered formalin, and processed for histopathology.
Study approvalAnimal procedures were performed in accordance with the rules of IACUC at the University of Florida. The primary human pancreatic cancer G-68 and G-46 cell lines and G192-p4, LM12-p3 and G176-p4 human pancreatic cancer PDXs were generated from deidentified tissues collected from patients. Informed written consent for the collection, and use of the tissues for research was obtained from all patients, and the use of these patient-derived cells and tissues for this study was approved by the University of Florida Institutional Review Board.
Statistical analysisFor analysis of the means of three or more groups, ANOVA tests were performed. In the event that ANOVA justified post hoc comparisons between group means, the comparisons were conducted using Tukey multiple-comparisons test. A two-sided unpaired Student t test was used for comparisons between the means of two groups. Apoptosis and spheroid assays statistical analyses were done by two-way ANOVA with Tukey post hoc test. A Kaplan–Meier test was used for the survival analysis, and the data were statistically analyzed using the log-rank (Mantel–Cox) test. P < 0.05 was considered to be statistically significant.
All other methods are described in Supplementary Information due to space limitation.
ResultsCRISPR screening with the RxG library found that targeting of BCL2L1 increases sensitivity of AsPC-1 cells to gemcitabine, 5FU, and niraparibWe constructed the RxG library, which targets 996 human target genes for which a pharmaceutical modulator had been identified (Supplementary Methods; Supplementary Tables S1 and S2). We carried out CRISPR screening in AsPC-1 pancreatic cancer cells using the RxG library to identify druggable genes that modulate sensitivity/resistance of AsPC-1 cells to gemcitabine, 5FU, and niraparib. (Fig. 1A). Briefly, AsPC-1 cells were transduced with the RxG library, puromycin selected (T0 PURO), and exposed to DMSO (vehicle), gemcitabine (3.2 μmol/L), 5FU (2.5 μmol/L), and niraparib (10 μmol/L) for 8 days in triplicate. Genomic DNA was isolated from each sample, sgRNA amplified, and the abundance of each sgRNA determined by next-generation sequencing. We assessed the depletion/enrichment of each gene in the treated samples as compared with the T0 PURO control using the MAGeCK algorithm (Fig. 1A; Supplementary Methods). Strikingly, targeted disruption of BCL2L1, which encodes BCL-XL, resulted in a significant reduction of AsPC-1 cells in gemcitabine-, 5FU-, and niraparib-treated samples but not in the DMSO-treated cells (Fig. 1B–D). These findings suggest that BCL-XL may enhance resistance to these agents in AsPC-1 cells. These results agree with previous findings, demonstrating that BCL-XL plays a crucial role in the development of chemoresistance in pancreatic cancer cells (5).
 Figure 1.
Figure 1. BCL2L1 (BCL-XL) provides resistance to gemcitabine treatment in pancreatic cancer. A, Representation of the Therapeutic Genome (RxG) CRISPR library screening to identify genes important for resistance to gemcitabine (GEM), 5FU, and niraparib (NIR) in the AsPC-1 pancreatic cancer cell line. B–D, Log2 volcano plots showing the results of the screening for gemcitabine (B), 5FU (C), or niraparib (D). Several biologically interesting hits (including BCL2L1) identified from the screening are highlighted in red. Each gene targeted by the library was ranked based on the Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) positive selection score. FDR, False discovery rate; FC, Fold change.
Expression of the BCL-2 family proteins in and sensitivity of pancreatic cancer cells to BH3 mimeticsTo gain more insight into the role of the BCL-2 family of proteins in the development of chemoresistance in pancreatic cancer cells, we analyzed the Cancer Cell Line Encyclopedia (CCLE) database and found that BCL2L1 mRNA is predominantly overexpressed in pancreatic cancer cell lines compared with the cell lines that were derived from other malignancies (Supplementary Fig. S1A). Similarly, when we evaluated the expression of the BCL-2 family of proteins in four commonly used pancreatic cancer cell lines (i.e., AsPC-1, BxPC-3, Mia PaCa-2, and PANC-1) and two newly generated patient-derived primary pancreatic cancer cell lines (i.e., G-46 and G-68; Supplementary Table S3; ref. 30), we found that BCL-XL and MCL-1 are abundantly expressed across all the pancreatic cancer cell lines, while BCL-2 expression was more variable (Supplementary Fig. S1B). Next, we determined the sensitivity of these pancreatic cancer cells to different BCL-2 family antiapoptotic protein inhibitors (commonly known as BH3 mimetics) including A1155463 (a BCL-XL-selective inhibitor), ABT199 (a BCL-2-selective inhibitor), S63845 (an MCL-1-selective inhibitor), ABT263 (a dual BCL-XL and BCL-2 inhibitor), and DT2216 (a BCL-XL specific PROTAC or degrader) as single agents as well as in different combinations. All of the pancreatic cancer cell lines tested were found to be largely resistant to these inhibitors when used individually (Supplementary Fig. S1C), suggesting that unlike many leukemia and lymphoma cells, pancreatic cancer cells are not dependent on single BCL-2 family members for survival. Next, we tested different combinations of the BH3 mimetics and found that all of these pancreatic cancer cell lines were resistant to the combination of A1155463 and ABT199 as well as S63845 plus ABT199, but were highly sensitive to the combination of S63845 with any of these BCL-XL–targeting agents including DT2216. However, MCL-1 inhibition causes severe toxicities including cardiotoxicity and hepatotoxicity, and these toxicities are exacerbated with dual targeting of MCL-1 and BCL-XL (33–35). Hence, to avoid these toxicities, alternative pharmacologic agents that could cause MCL-1 suppression selectively in tumor cells would be of great importance to develop highly effective combination therapies with a platelet-sparing BCL-XL–targeting agent such as DT2216 (23, 26).
A combination of gemcitabine and DT2216 synergistically kills pancreatic cancer cells in vitroThe priming of cancer cells to apoptosis with chemotherapeutic drugs has been shown to significantly increase the effectiveness of BCL-2 family inhibitors. In our RxG CRISPR screening, we identified that BCL-XL plays a crucial role in the development of gemcitabine resistance. Thus, to improve the efficacy of gemcitabine, we investigated the potential to use the combination of gemcitabine and DT2216 for the treatment of pancreatic cancer.
First, we determined the effect of gemcitabine and DT2216 as single agents and in combination against different pancreatic cancer cell lines. Flow-cytometric analysis clearly showed that the combination of gemcitabine and DT2216 significantly increased apoptosis compared with the individual agents in four commonly used pancreatic cancer cell lines (AsPC-1, BxPC-3, Mia PaCa-2, and PANC-1; Fig. 2A, Supplementary Fig. S2A) as well as two newly generated patient-derived primary pancreatic cancer cell lines (G-68 and G-46; Fig. 2B; Supplementary Fig. S2B and S3A, B). We observed that, although, basal levels of BCL-XL protein expression were different among the tested cell lines, all the cell lines were found to be sensitive to the combination of gemcitabine and DT2216, and that of gemcitabine and S63845 (Fig. 2A and B; Supplementary Fig. S1C), indicating that the efficacy of DT2216 may be independent of the basal expression of BCL-XL. Furthermore, the combination of gemcitabine and DT2216 was found to synergistically reduce viability, colony formation, and tumor spheroid growth in the G-68 and G-46 cell lines (Fig. 2C–G; Supplementary Fig. S3C–S3G). These data confirm a synergistic interaction between gemcitabine and DT2216 against pancreatic cancer cells in vitro.
 Figure 2.
Figure 2. The combination of DT2216 and gemcitabine synergistically kills pancreatic cancer cells in vitro. A, Percentage of total apoptotic cells (the sum of early [Annexin V+] and late [PI+/Annexin V+] apoptotic cells) after the cells were treated with vehicle (Veh) or with the indicated concentrations of DT2216 (DT) or gemcitabine (GEM) alone or in combination for 72 hours. Data are presented as the mean ± SD of four independent experiments for each cell line and condition. a, b and c, P < 0.05 versus. Veh, DT and GEM, respectively. B, Percentage of total apoptotic G-68 primary pancreatic cancer cells after they were treated with vehicle (Veh) or the indicated concentrations of DT or gemcitabine alone or in combinations for 72 hours. Data are presented as mean ± SD (n = 3 independent experiments). *, P < 0.05; ****, P < 0.0001. Representative flow cytometric quadruple graphs for A and B are shown in Supplementary Fig. S2A and S2B, respectively. C, Percentage viability of G-68 cells after they were treated with increasing concentrations of DT, gemcitabine, or their combination (GEM+DT; 1:1 ratio) for 72 hours. The table shows the IC50 values of DT, gemcitabine, or their combination (GEM+DT). IC50 values are shown for a representative experiment out of three independent experiments. The CI value (< 1) indicates a synergy between DT and gemcitabine. D, G-68 cells were treated with the indicated concentrations of DT or gemcitabine alone or in combination for 72 hours followed by incubation in drug-free medium for another two weeks. Crystal violet staining was performed to visualize the colonies. Data are representative images from two independent experiments. E, G-68 cells were seeded in a 48-well plate and allowed 48 hours for spheroid formation before DT and/or gemcitabine treatment. Micrographs (magnification at 50×) of spheroids in the corresponding wells after they were treated with the indicated concentrations of DT or gemcitabine alone or in combination for 10 days are shown. F, Microphotographs (magnification 50×) of spheroids from the marked areas in E. G, Quantification of the number of spheroids from E as a percentage of Veh. a, b and c, P < 0.05 versus Veh, DT, and GEM, respectively. Data presented in E–G are representative of three independent experiments. Statistical significance in A, B, and G was determined by two-way ANOVA with Tukey post hoc test.
BCL-XL degradation by DT2216 increases MCL-1 dependence and sensitivity to gemcitabine in pancreatic cancer cellsHaving established the synergistic activity of gemcitabine and DT2216 against pancreatic cancer cells in vitro, we next sought to uncover the molecular mechanisms behind their synergy. We chose the G-68 cell line for these mechanistic studies because it is a primary pancreatic cancer cell line from a patient with a T3N1 tumor harboring both the KRASG12D and TP53R248W mutations. It resembles invasive pancreatic cancer in patients more than the commonly used human pancreatic cancer cell lines (30). DT2216 potently degraded BCL-XL in a concentration-dependent manner without any significant effect on BCL-2 and MCL-1 (Fig. 3A). On the other hand, gemcitabine treatment significantly downregulated MCL-1 protein expression and partially downregulated BCL-XL in a concentration-dependent manner, while BCL-2 levels were not affected. In addition, gemcitabine also increased the expression of NOXA that selectively binds MCL-1 to induce apoptosis (Fig. 3B; ref. 36). Next, we analyzed the effect of the gemcitabine-DT2216 combination on BCL-2 family members and apoptosis markers. We found that the combination reduced the levels of MCL-1 and BCL-XL in the cells. Furthermore, apoptosis markers such as cleaved-caspase 3 and cleaved-PARP levels were significantly increased with the combination treatment compared with treatment with the individual agents (Fig. 3C). This enhanced effectiveness of the combination treatment further supports the codependency of these cells on BCL-XL and MCL-1. Next, we determined whether differential binding of BIM to BCL-XL and MCL-1 was the reason for the limited single-agent activity in pancreatic cancer cells. To directly compare the binding of BIM to BCL-XL and MCL-1, we performed immunoprecipitation of BCL-XL and MCL-1. We observed that at the baseline, BIM is more associated with BCL-XL, whereas degradation of BCL-XL by DT2216 led to its increased association with MCL-1 (Fig. 3D, E). This confirms that BCL-XL degradation by DT2216 increases MCL-1 dependence and could be a reason for the limited activity of a single agent. Together, downregulation of MCL-1 by gemcitabine under DT2216 treatment confirmed the mechanism behind the combination effect. The codependency of pancreatic cancer cells on BCL-XL and MCL-1 was further validated using cell viability assays utilizing an MCL-1-specific inhibitor (S63845) and DT2216 (Fig. 3F and Supplementary Fig. S1C). Collectively, our data suggest that gemcitabine-induced MCL-1 downregulation with BCL-XL degradation by DT2216 may be primarily responsible for this synthetic lethality in pancreatic cancer cells. Furthermore, RT-PCR analysis showed that the altered protein expression of BCL-2 family members in the combination treatment is not associated with changes in mRNA levels (Fig. 3G and Supplementary Table S4).
 Figure 3.
Figure 3. Synergy between DT2216 and gemcitabine attributes to a combined depletion of BCL-XL and MCL-1. A, Immunoblot analysis of BCL-XL, BCL-2, and MCL-1 in G-68 cells after they were treated with the indicated concentrations of DT2216 (DT) for 16 hours. B, Immunoblot analysis of BCL-XL, BCL-2, MCL-1, and NOXA in G-68 cells after they were treated with the indicated concentrations of gemcitabine (GEM) for 48 hours. C, Immunoblot analysis of BCL-XL, BCL-2, MCL-1, NOXA, and apoptosis markers – cleaved (C) and full-length caspase-3 and PARP in G-68 cells after they were treated with the indicated concentrations of gemcitabine and/or DT for 48 hours. Immunoblots presented in A-C are representative of three independent experiments. D, G-68 cells were treated with gemcitabine (1 μmol/L) and/or DT (0.1 μmol/L) for 48 hours. Cell lysates were subjected to co-immunoprecipitation (Co-IP) using BCL-XL or MCL-1 antibodies followed by immunoblotting to detect BCL-XL, MCL-1, and BIM. E, The levels of BCL-XL, MCL-1, and BIM in the inputs for D. β-actin was used as an equal loading control for all immunoblot analyses presented in A–E. F, Viability of G-68 cells after they were treated with increasing concentrations of DT or S63845 (S) individually or in combination (DT+S) for 72 hours. IC50 values are shown in the table. The CI value indicates a synergy between DT and S. G, G-68 cells were treated with 0.1 μmol/L of DT or 1 μmol/L of gemcitabine alone or in combination (gemcitabine + DT) for 48 hours. The fold changes in the expressions of BCL2L1 (encodes BCL-XL), BCL2, MCL1, and PMAIP1 (encodes NOXA) mRNA are shown. GAPDH was used as an internal control. Data are from a single experiment performed in triplicate (mean ± SD).
Combination of gemcitabine and DT2216 can more effectively suppress the growth of G-68 xenografts and PDX tumors than either agent alone in miceGiven the excellent synergetic activity in pancreatic cancer cells in vitro, we sought to evaluate the effect of gemcitabine and DT2216 in vivo. Xenograft tumor models were established using the G-68 cell line. Tumor-bearing NOD-scid IL2Rgnull (NSG) mice were randomized into four groups (n = 7 mice per group) once the tumors reached 100 to 200 mm3. The animals were treated intraperitoneally (i.p.) with vehicle, gemcitabine (20 mg/kg, every 7 days), DT2216 (15 mg/kg, every 4 days), or the combination of gemcitabine and DT2216 (Fig. 4A). In this model, gemcitabine was slightly more effective than DT2216 in inhibiting tumor growth and increasing the survival of the tumor-bearing mice, whereas the combination treatment was more effective than either agent alone without any significant decrease in body weight (Fig. 4B–E). Previous reports have shown that a combination of BCL-XL and MCL-1 inhibitors induced acute hepatotoxicity in mice (34). Thus, we studied the histopathology of the liver, heart, and kidney in mice receiving the individual treatments or the combination therapy and found no evidence of any organ toxicity, which is in agreement with the observation that there was no body weight change during the treatments (Supplementary Fig. S4A).
 Figure 4.
Figure 4. DT2216 increased the antitumor efficacy of gemcitabine in a patient-derived G-68 pancreatic cancer cell xenograft model. A, Representation of the experimental design of the G-68 xenograft study. Tumor-bearing mice were administered Veh, DT2216 (DT), gemcitabine (GEM), or a combination of DT and gemcitabine at the indicated dosing regimen. B, Change in body weight during the course of treatment. C, Graph showing the tumor volume changes in each group after the start of treatment until the control animals were euthanized. Data are presented as mean ± SEM (n = 7 mice in each group at the start of treatment). Statistical significance was determined by unpaired two-sided Student t test test. **, P < 0.01; ****, P < 0.0001. D, Kaplan–Meier survival analysis with medium survival time of mice in each group. Statistical significance was determined by Mantel–Cox test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. E, Representative H&E staining images of tumors in each treatment group at 200x magnification, scale bar = 50 μm.
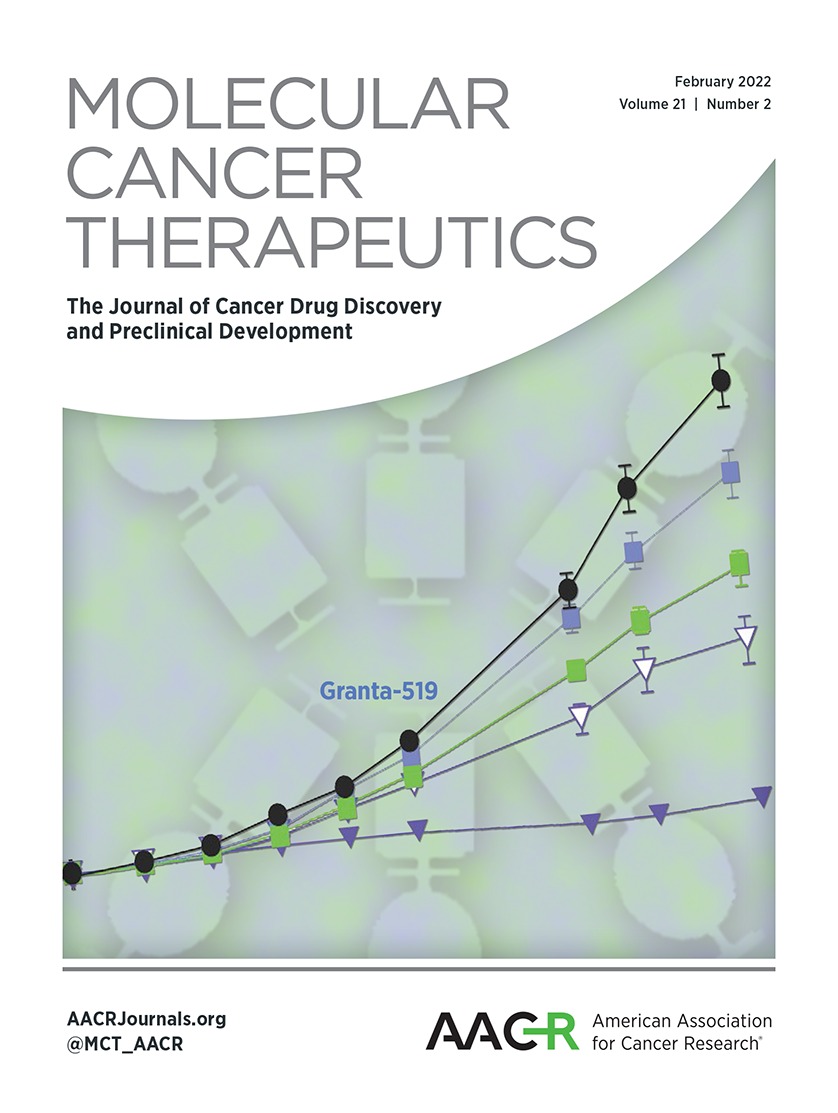
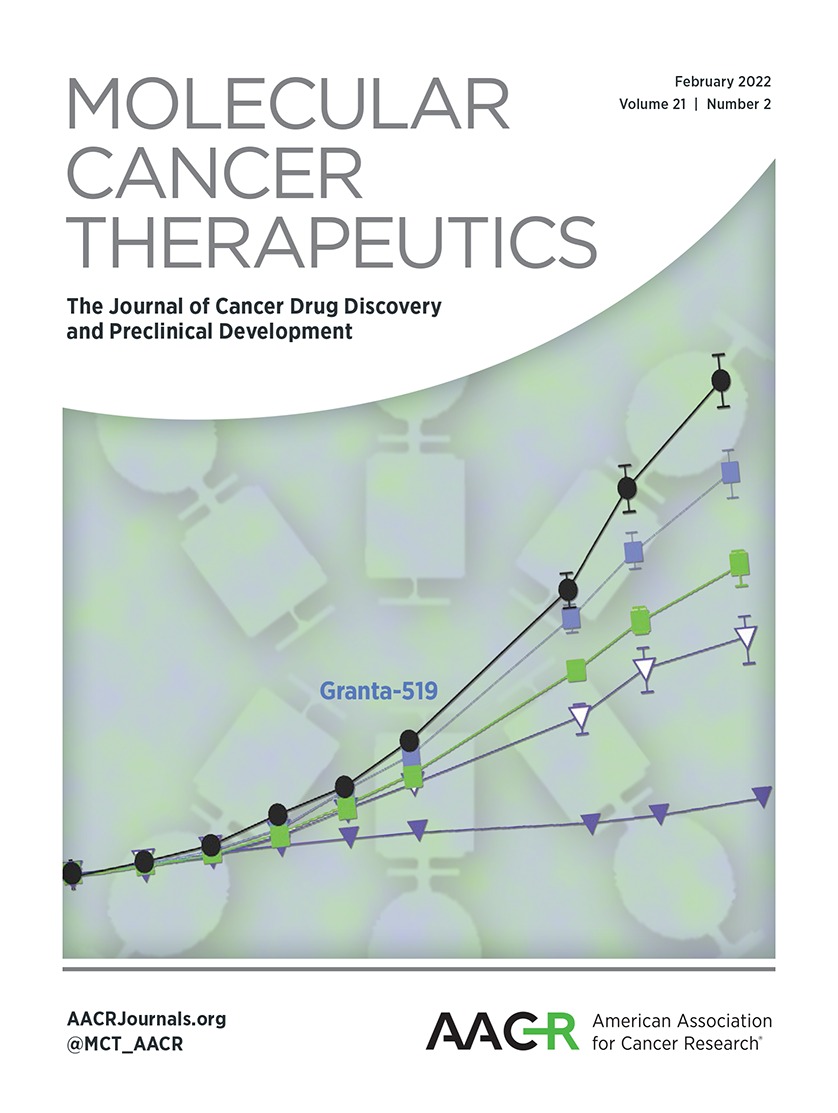
Recently, it has been shown that PDX tumor models can better recapitulate tumor biology, stromal content, genetic mutations, and heterogeneity of human diseases than conventional tumor xenograft models employing cancer cell lines (37). They are also more predictive of clinical outcomes of experimental therapeutic agents than the latter (37). We have established a number of pancreatic cancer PDX models and selected G192-p4, G176-p4, and LM12-p3 human pancreatic cancer PDX models to further evaluate the therapeutic efficacy of gemcitabine and DT2216 alone or in combination (Fig. 5A; Supplementary Table S5; refs. 32, 38, 39). In the G192-p4 PDX model, the combination treatment significantly inhibited tumor growth compared with monotherapy of gemcitabine or DT2216 (Fig. 5B). In addition, histopathology from the G192-p4 PDX model showed a significant decrease in tumor burden in mice treated with the combination, and no evidence of organ toxicity was observed in the liver, kidney, or heart (Fig. 5C and Supplementary Fig. S4B). The combination of gemcitabine and DT2216 was also more effective in inhibiting tumor growth in the G176-p4 model than either agent alone (Fig. 5D). In LM12-p3 PDX, the combination treatment caused more reduction in tumor volume than either treatment alone, but the difference was not statistically significant (Fig. 5E). Interestingly, DT2216 was equally effective in degrading BCL-XL in tumors harvested from all these PDXs (Supplementary Fig. S5A), which suggests that the differential efficacy of the combination against these PDXs may not be attributed to differential BCL-XL degradation. In fact, the efficacy of the combination appears to be correlated with MCL-1 expression in these PDXs, that is, PDXs with high MCL-1 expression (G192-p4) responded best to the treatment (Supplementary Fig. S5B). In addition, the heterogeneity of the PDXs may also contribute to the differences of their responses to the combination therapy. In fact, LM12-p3 PDX tumor tissues were derived from an aggressive liver metastasis, and thus appeared more resistant to any of these treatments than the other two PDXs (Fig. 5). Together, these data illustrate that the combination of gemcitabine and DT2216 is potentially a more effective treatment for some pancreatic cancers than gemcitabine alone. Future studies will be needed to identify biomarkers that can be used to stratify pancreatic cancer patients for this combination therapy.
 Figure 5.
Figure 5. DT2216 increased the antitumor efficacy of gemcitabine in pancreatic cancer PDX models. A, Representation of the experimental design of the PDX studies. Tumor-bearing mice were administered Veh, DT2216 (DT), gemcitabine (GEM), or a combination of DT and gemcitabine at the indicated dosing regimen. B, Graph showing the tumor volume changes in G192-p4 PDXs in each group after the start of treatment. C, Representative H&E staining images of G192-p4 PDX tumors in each treatment group at 200 × magnification, scale bar = 50 μm. D and E, Graphs showing the tumor volume changes in G176-p4 (D) and LM12-p3 (E) PDXs in each group after the start of treatment. The data presented in (B), (D) and E are mean ± SEM (n = 7 mice in each group at the start of treatment). Statistical significance was determined by unpaired two-sided Student t test. *, P < 0.05; **, P < 0.01; ns, not significant.
DiscussionPancreatic cancer is a difficult-to-treat cancer with gemcitabine as a current first-line standard of care. Unfortunately, gemcitabine is ineffective to treat this deadly disease because of development of drug resistance (4–7). The current studies emanate from the fact that BCL-XL is the most highly expressed gene in pancreatic cancer as analyzed through the CCLE transcriptomics database. We found that compared to BCL-2, the expression of BCL-XL is high in all the pancreatic cancer cells examined in our study. The importance of BCL-XL in the progression of PanIN to PDAC has also been demonstrated by others in pancreas-specific KrasG12D (P-KrasG12D) mice (12). A recent study showed that ABT263 increased the antitumor effect of prexasertib, a Chk1 inhibitor, by inducing apoptosis in pancreatic cancer cells via inhibition of BCL-XL but not BCL-2 (40). Furthermore, Zhang Z and colleagues showed that GATA1 induced gemcitabine resistance in pancreatic cancer cells through the upregulation of BCL-XL expression (41). Using the RxG CRISPR screening, we identified BCL-XL as the most common gene providing resistance to not only gemcitabine, but also to other chemotherapeutics such as 5FU and NIR. These findings confirm that BCL-XL is involved in the development of therapeutic resistance in pancreatic cancer and is a potential key druggable pancreatic cancer target.
Since on-target thrombocytopenia is a major hurdle in the clinical development of BCL-XL inhibitors, we recently adopted a novel PROTAC technology to overcome this toxicity by converting ABT263 (BCL-XL/2 dual inhibitor) into a BCL-XL selective PROTAC, named DT2216 (20–22). DT2216 is currently in phase I clinical studies for patients with relapsed/refractory malignancies (NCT04886622). In the current study, through a series of in vitro assays and in vivo studies including the use of our newly established patient-derived primary pancreatic cancer cells lines and PDX models, we confirmed that DT2216 can sensitize pancreatic cancer cells to gemcitabine. These results suggest that the gemcitabine–DT2216 combination is a promising new therapy against pancreatic cancer, which will be useful to guide the clinical development of DT2216.
The mechanism by which gemcitabine and DT2216 can synergistically kill pancreatic cancer cells is because they can simultaneously target MCL-1 and BCL-XL. Pancreatic cancer cells express high levels of not only BCL-XL but also MCL-1 and are thus insensitive to individual inhibition of BCL-XL or MCL-1. Therefore, like many other solid tumor cells, pancreatic cancer cells respond poorly to BCL-2 family inhibitors when used as a single ag
Comments (0)