
Remember me



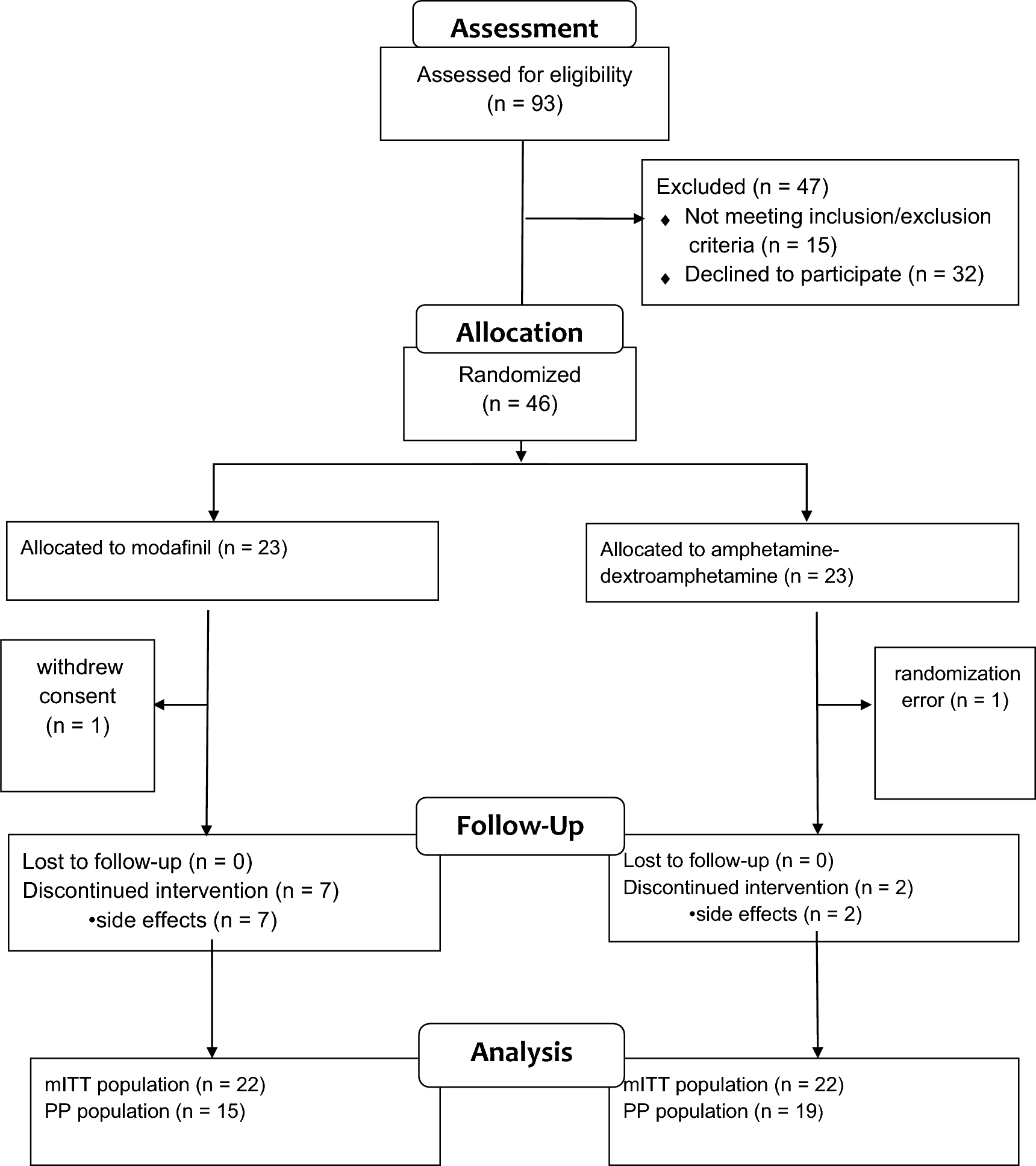






Between 4 October 2018 and 13 April 2021, a total of 298 patients (rollover, n = 246; de novo, n = 52) were screened (Fig. 1). Of these, 247 patients (rollover patients, n = 216; de novo patients, n = 31) were included in the safety analysis set/efficacy analysis set. Of these, 138 (55.9%) patients (rollover patients, n = 132; de novo patients, n = 6) completed the study, and 109 (44.1%) patients (rollover patients, n = 84; de novo patients, n = 25) discontinued the study. The most common reasons for discontinuation were adverse events [n = 66 (26.7%)], followed by patient withdrawal [n = 25 (10.1%)], and protocol deviation [n = 8 (3.2%)].
Fig. 1
Participant flow chart. BRX brexpiprazole, LTE long-term extension, PBO placebo
Baseline demographics and clinical characteristics are presented in Table 1. Mean (SD) age was 44.4 (13.9) years, and mean (SD) MADRS, HAM-D 17, and CGI-S scores at baseline were 20.1 (8.9), 15.7 (6.1), and 3.6 (0.9), respectively.
Table 1 Patient demographics and clinical characteristics (safety analysis set)Baseline demographic and clinical characteristics were similar between rollover patients and de novo patients, with a few notable differences. Given that de novo patients were eligible if they were aged ≥ 65 years, the mean age was predictably lower in the rollover group [40.7 (10.3) years] compared with the de novo group [70.3 (4.5) years]. The mean (SD) duration of the first episode of MDD was also longer in de novo patients [90.7 (85.5) months versus 69.5 (70.6) months, respectively], and more patients had a recurrent episode of MDD (77.4% versus 64.4%, respectively) compared with rollover patients.
The adherence rate was ≥ 80% in all patients, and 99.2% (n = 245) achieved ≥ 90% adherence. In the safety/efficacy analysis set (n = 247), more than half [55.9% (n = 138)] of patients received brexpiprazole for at least 337 days. Of the 31 de novo patients, 19.4% (n = 6) received brexpiprazole for at least 337 days, and 51.6% (n = 16) received brexpiprazole for less than 85 days.
3.1 SafetyTEAEs of any grade occurred in 93.5% (n = 231) of patients receiving brexpiprazole, with no notable differences between rollover [93.1% (n = 201)] and de novo [96.8% (n = 30)] patients.
The incidence of common (incidence ≥ 5%) TEAEs are presented in Table 2. The most common (≥ 5% of patients) TEAEs of any grade with brexpiprazole were weight gain [33.2% (n = 82)], akathisia [23.5% (n = 58)], nasopharyngitis [21.1% (n = 52)], somnolence [10.5% (n = 26)], insomnia [9.7% (n = 24)], headache [9.3% (n = 23)], tremor [8.5% (n = 21)], increased appetite [7.3% (n = 18)], extrapyramidal disorder [6.5% (n = 16)], malaise [6.1% (n = 15)], and constipation and major depression [5.7% (n = 14) each]. TEAEs reported in ≥ 10% of de novo patients were akathisia [38.7% (n = 12)], tremor [19.4% (n = 6)], nasopharyngitis [16.1% (n = 5)], and weight gain, dyskinesia, extrapyramidal disorder, and major depression [12.9% (n = 4) each]. The discontinuation rate due to TEAEs was higher in de novo patients compared with rollover patients, and the incidences of akathisia, tremor, and extrapyramidal disorder were higher in de novo patients. An examination of adverse events (AEs) by antidepressant type (SSRIs, SNRIs, and mirtazapine) revealed no major differences in the incidence of AEs between groups (Supplementary Table S2).
Table 2 Frequency of common (≥ 5%) TEAEs occurring in any group during the study period (safety analysis set)Most TEAEs were mild to moderate in severity in both the overall brexpiprazole group and the de novo group aged ≥ 65 years. Severe TEAEs in the safety analysis set (n = 247) were infrequent [4.9% (n = 12)], but were more common in de novo patients [12.9% (n = 4)], although the sample size was small (n = 31).
Overall, most TEAEs occurred by Day 56 of treatment, with a peak in incidence observed from Day 29 to 56 of treatment [19.2% (n = 45)], ranging from 0% to 7.3% thereafter. All TEAEs in de novo patients occurred by Day 112 of treatment (except for in one patient in whom the first TEAE occurred after the end of treatment), and the TEAE incidence was highest from Day 15 to 28 of treatment [36.7% (n = 11)]. There was no increase in TEAE incidence with increasing treatment duration and no significant difference in the time to first onset of TEAE incidence between all brexpiprazole patients and de novo patients.
Serious adverse events (SAEs) occurred in 3.6% (n = 9) of patients receiving brexpiprazole [rollover, 3.7% (n = 8); de novo, 3.2% (n = 1)], none of which occurred in more than two patients. Serious adverse events included appendicitis, arterial occlusive disease, colon cancer, death, depression, duodenal perforation, dyskinesia, human immunodeficiency virus (HIV) infection, ligament sprain, lung neoplasm malignant, major depression, metastases to lymph nodes, and septic shock [0.4% (n = 1) each]. The incidence of SAEs considered by the investigator as related to brexpiprazole was 0.5% (n = 1) in rollover patients and 3.2% (n = 1) in de novo patients, and included dyskinesia in one patient who rolled over from the placebo group and major depression in one patient in the de novo group. During the open-label treatment phase, suicidal ideation was reported in 9.7% of patients by C-SSRS evaluation, but there were no new reports of suicidal behavior or suicidal ideation as a TEAE. One death considered unrelated to study treatment was reported in a 49-year-old male who died of an unknown cause after study completion (date of death unknown).
In total, 26.7% (n = 66) of patients discontinued brexpiprazole treatment due to TEAEs; discontinuations due to TEAEs were more common in de novo patients compared with rollover patients [58.1% (n = 18) versus 22.2% (n = 48), respectively]. The most common (≥ 5 patients) TEAEs leading to treatment discontinuation were akathisia [n = 10 (de novo patients, n = 6)]; weight gain [n = 7 (de novo patients, n = 1)]; malaise, extrapyramidal disorder, and major depression [n = 5 (de novo patients, n = 2) each]; and somnolence [n = 5 (de novo patients, n = 0)]. Of these, all TEAEs were considered related to brexpiprazole, with the exception of major depression [three out of five cases (de novo patient, n = 1)]. Of the events leading to brexpiprazole discontinuation, most events that occurred in ≥ 5 patients receiving brexpiprazole were resolved or resolving.
Extrapyramidal symptom-related TEAEs occurred in 39.7% (n = 98) of patients, with a higher incidence observed in de novo patients [71.0% (n = 22)] than rollover patients [35.2% (n = 76)]. The most common (≥ 5% of patients) extrapyramidal-symptom-related TEAEs occurring in rollover patients were akathisia [21.3% (n = 46)], tremor [6.9% (n = 15)], and extrapyramidal disorder [5.6% (n = 12)]. The most common (≥ 5% of patients) extrapyramidal-symptom-related TEAEs occurring in de novo patients were akathisia [38.7% (n = 12)], tremor [19.4% (n = 6)], dyskinesia and extrapyramidal disorder [12.9% (n = 4) each], bradykinesia and dystonia [9.7% (n = 3) each], and parkinsonism [6.5% (n = 2)]. Dyskinesia events occurred in 4.5% (n = 11) of patients overall, including 2.1% (n = 3) of rollover patients previously receiving brexpiprazole and 5.4% (n = 4) of rollover patients previously receiving placebo. No events of tardive dyskinesia were reported.
The mean (SD) increase in body weight from baseline to Week 24 was 3.1 (4.6) kg (n = 167; OC) and from baseline to Week 52 was 4.2 (6.5) kg (n = 138; OC) and 3.3 (5.5) kg (n = 246; LOCF) in patients receiving brexpiprazole, and 0.2 (1.9) kg (n = 7) and 0.1 (3.7; n = 6; OC) and 0.3 (2.2) kg (n = 31; LOCF), respectively, in de novo patients (Fig. 2). Increase in body weight ≥ 7% at any postbaseline visit occurred in 44.5% (n = 110) of patients receiving brexpiprazole and 9.7% (n = 3) of de novo patients, and a decrease in body weight of ≥ 7% at any postbaseline visit occurred in 5.7% (n = 14) of patients receiving brexpiprazole and 9.7% (n = 3) of de novo patients (Supplementary Table S3). Of the patients who experienced a weight gain of ≥ 7%, 72.7% (n = 80) of those had a baseline BMI of < 25 kg/m2.
Fig. 2
Mean change in body weight from baseline over the 52-week study period
There were no clinically relevant findings with respect to events related to electrocardiogram findings, or prolactin, blood biochemistry and hematology, lipids, or glucose, including the incidence of shifts to abnormal levels (see Supplementary Table S3, which summarizes additional safety outcomes). Abnormal electrocardiogram findings were reported in two patients and included QT prolonged [0.5% (n = 1)] and T wave inversion [3.2% (n = 1)], in a patient each in the rollover group and de novo group, respectively. In patients receiving brexpiprazole, the mean (SD) prolactin level changes from baseline to Week 24 (OC) were 4.8 (24.7) μg/L (n = 61) in females and 0.2 (3.7) μg/L (n = 106) in males, and those from baseline to Week 52 (OC) were 1.8 (24.0) μg/L (n = 55) in females and − 0.2 (4.2) μg/L (n = 83) in males. In de novo patients, mean (SD) prolactin level changes were similar between males and females.
The mean change in fasting glucose from baseline to Week 24 (OC) was 2.3 (9.0) mg/dL (n = 110), and that from baseline to Week 52 (OC) was 2.6 (10.4) mg/dL (n = 137). The proportion of patients who met central obesity criteria and two out of three other criteria for treatment-emergent metabolic syndrome at any visit was 2.7% (6 of 226 patients who did not meet the criteria at baseline and who had a postbaseline measurement) in patients receiving brexpiprazole and 3.2% (1 of 31 patients who did not meet the criteria at baseline and who had a postbaseline measurement) in de novo patients. No clinically meaningful changes were observed on formal extrapyramidal syndrome rating scales (see Supplementary Table S4, which provides DIEPSS, AIMS, and BARS score changes) after 52 weeks, with the exception of the DIEPSS.
3.2 EfficacyImprovements in the MADRS total score were observed from baseline over the course of the 52-week study for all patients receiving brexpiprazole, including both rollover and de novo patients (Fig. 3). Improvements in other efficacy endpoints, including CGI-S, HAM-D 17 item total score, and SDS mean score were also observed from baseline over the 52-week study (Table 3), indicating a sustained improvement in symptoms with long-term brexpiprazole treatment. Similar results were observed in de novo patients, with improvements from baseline also observed for each endpoint in patients aged ≥ 65 years.
Fig. 3
Mean change in MADRS total score from baseline over the 52-week study period. MADRS Montgomery–Åsberg Depression Rating Scale
Table 3 Summary of efficacy and other outcomes at Weeks 24 (OC) and 56 (OC and LOCF) of the study period (efficacy analysis set)The MADRS response rate, MADRS remission rate, and CGI-I rate were maintained throughout the treatment period for all patients receiving brexpiprazole, including de novo patients (Table 3).
The mean (SD) change from baseline in MADRS total score over the course of the study is presented in Fig. 3. The mean (SD) change from baseline in MADRS total score in patients receiving brexpiprazole was − 5.0 (8.2; n = 167) at Week 24 (OC) and − 7.3 (8.7; n = 138) at Week 52 (OC); with LOCF, the mean (SD) change from baseline in MADRS total score at Week 52 was − 4.6 (9.2; n = 247). Except for mirtazapine [only de novo (older adult) patients were eligible for administration], which was discontinued in all patients, changes in MADRS from baseline to 24 weeks (OC), 52 weeks (OC), and 52 weeks (LOCF) were examined by SSRI and SNRI, and no major differences were observed (Supplementary Table S5). The overall MADRS response rate in patients receiving brexpiprazole was 25.1% (n = 42) at Week 24 (OC), 41.3% (n = 57) at Week 52 (OC), and 29.1% (n = 72) at Week 52 (LOCF). The overall MADRS remission rate in patients receiving brexpiprazole was 19.8% (n = 33) at Week 24 (OC), 34.8% (n = 48) at Week 52 (OC), and 25.5% (n = 63) at Week 52 (LOCF). The CGI-I rate in patients receiving brexpiprazole was 32.3% (n = 54) at Week 24 (OC), 50.0% (n = 69) at Week 52 (OC), and 36.0% (n = 89) at Week 52 (LOCF). Similar findings were observed in de novo patients, although sample sizes were small (Table 3).
The mean (SD) SDS score was 4.8 (2.4) at baseline in all patients receiving brexpiprazole. The mean (SD) change from baseline in SDS score was − 0.6 (1.9; n = 167) at Week 24 (OC), − 1.0 (2.2; n = 138) at Week 52 (OC), and − 0.5 (2.4; n = 229) at Week 52 (LOCF).
Comments (0)