Cell culture and treatment
PC12 cells (CRL-1721™, ATCC, Manassas, VA, USA) were maintained in Dulbecco’s Modified Eagle’s medium (DMEM) (Gibco, Grand Island, NY, USA) supplemented with 10% FBS (Gibco) and 1% Penicillin-Streptomycin (10,000 U/mL) (Gibco) at 37 °C in a humidified incubator with 5% carbon dioxide. For lidocaine stimulation, PC12 cells (P9-13 passage) were treated with different concentrations of lidocaine (0, 0.25mM, 0.5mM, 1mM). For DEX treatment, PC12 cells were treated with 1mM lidocaine combined with different concentrations of DEX (0, 10 µΜ, 25µΜ, 50 µΜ).
Cell transfection
PC12 cells (1 × 105) were cultured until reaching 70–80% confluence in 12-well plates. Small interference RNA (siRNA) targeting CISD2 (si-CISD2) and its negative control (si-NC) were transfected into PC12 cells using Lipofectamine 2000 (Invitrogen).
MTT assay
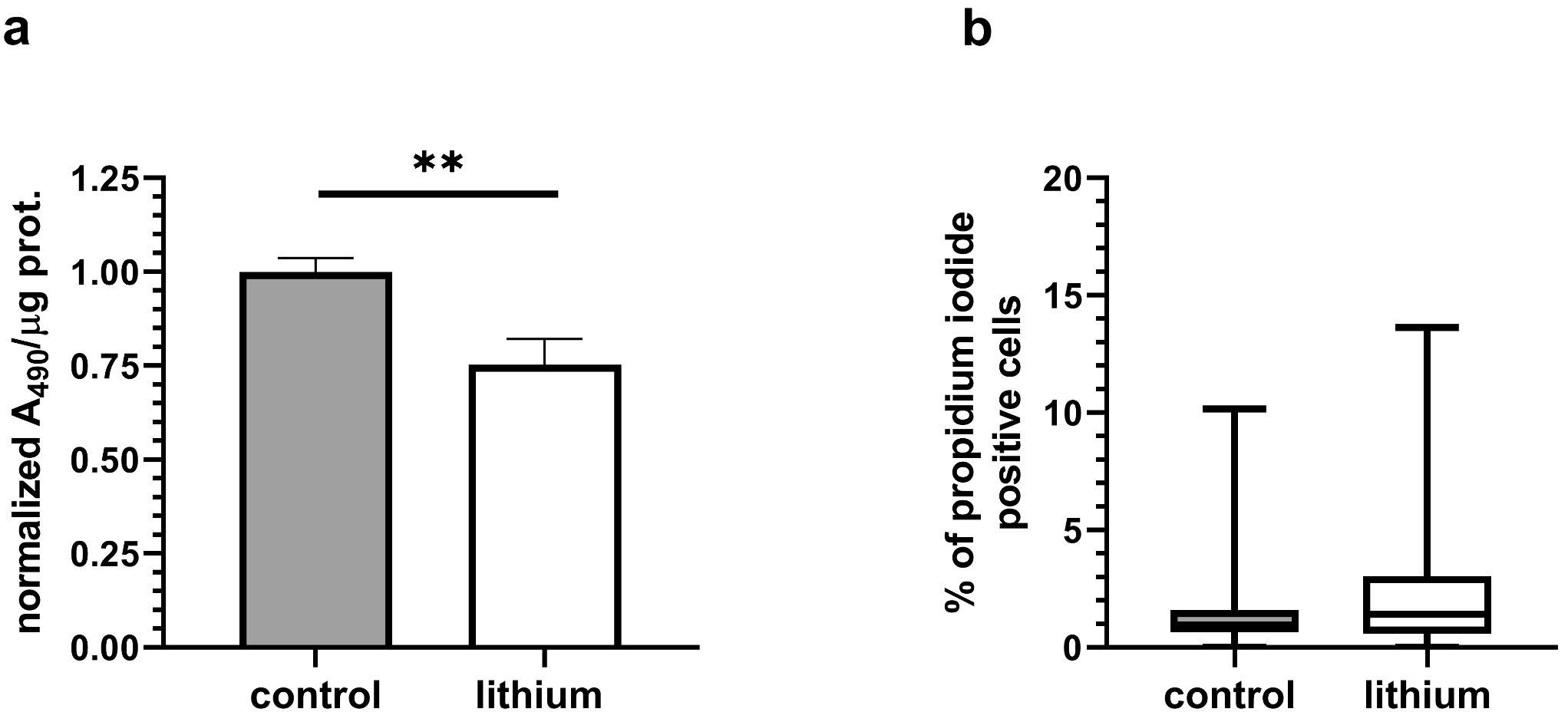
Cell viability was determined by using the MTT assay kit (M1020, Solarbio, Beijing, China). PC12 cells were inoculated at a density of 1 × 104 cells per well and incubated for 12 h. Subsequently, medication was used to treat and incubate for 24 h. Ten microliters of MTT solution were added and cultured for 4 h. The formazan solution (110 µL) was added to each well to dissolve the crystals after removing the supernatant. Measurement of the absorbance values of each well at 490 nm was conducted on an enzyme-linked immunosorbent assay (ELISA).
Flow cytometry for ROS measurement in cells
A reagent kit for detecting reactive oxygen species using fluorescent probe DCFH-DA (Beyotime, China) was used in this experiment. Dilution of DCFH-DA was performed using a serum-free culture medium at a ratio of 1:1000, resulting in a final concentration of 10 µmol/L. The collected cells were suspended in diluted DCFH-DA at a cell concentration of 1 × 106/mL. To ensure sufficient contact between the probe and the cell, mixing was performed by inverting it up and down every 3–5 min. After incubation for 20 min, the cells were washed three times using a serum-free cell culture medium. The detection of fluorescence intensity was performed by flow cytometry (Biosciences, San Diego, CA, USA) using 488 nm excitation wavelength and 525 nm emission wavelength.
Detection of malondialdehyde (MDA), glutathione (GSH) and Fe2+
The levels of malondialdehyde (MDA), GSH, and ferrous iron (Fe 2+) in PC12 cells and/or spinal cord lysate were detected by the MDA assay kit (S0131, Beyotime), GSH assay kit (S0053, Beyotime) and iron assay kit (MAK025, Sigma, USA) according to relevant manufacturers’ protocols.
LysoTracker staining
The cell culture medium was removed, followed by the addition of LysoTracker Green staining solution (C1047S, Beyotime) with a final concentration of 50–75 nM and preheated at 37℃. After incubation for 30 min, the Lyso Tracker Green staining solution was removed, and a fresh cell culture medium was added. Subsequently, the fluorescence intensity of lysosomes was observed using a fluorescence microscope (TE-2000, Nikon, Tokyo, Japan).
Immunofluorescence staining
P12 cells in each group were cultured and permeated by 0.03% Triton X-100 (Beyotime). After incubation for 60 min, the cells were fixed, followed by blocking with 0.1% BSA (Beyotime). Subsequently, the primary antibodies anti-ferritin (ab75973, Abcam, Cambridge, UK) and anti-microtubule-associated protein 1 light chain 3 (LC-3) were applied to the medium. The cells were analyzed via fluorescence microscopy (Tokyo).
Western blotting
Total protein was extracted using a RIPA buffer (Solarbio) and then measured using a commercial BCA kit (Pierce, Rockford, IL, USA). Equal amounts of protein (30 µg) were then separated by 10% SDS-PAGE, transferred to polyvinylidene fluoride membranes, and blocked into 5% fat-free milk. After incubation with primary antibodies LC3I/II (1:1000; #4108, Cell Signaling Technology, Beverly, MA, USA), sequestosome 1 (p62,1:10000; ab109012, Abcam), glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:1000; ab181602, Abcam), CISD2 (1:1000; #60758, Cell Signaling Technology), ferritin heavy chain 1 (FTH1,1:1000; #4393, Cell Signaling Technology), solute carrier family 7-member 11-glutathione (SLC7A11, 1:1000; ab307601, Abcam), GPX4 (1:1000; ab181602, Abcam) and nuclear receptor coactivator 4 (NCOA4) (1:1000; #66849, Cell Signaling Technology), the membranes were incubated with secondary antibody conjugated to horseradish peroxidase. After enhanced chemiluminescence (ECL) detection, protein bands were quantified using ImageJ version 1.51 software (NIH).
SCI and treatment of drugs
Healthy Sprague Dawley (SD) rats (male, adult, 230–270 g) (Experimental Animal Center of the Academy of Military Medical Sciences, Beijing, China) were selected. All rats were provided ad libitum access to food and clean water. The indoor temperature of the artificial control room was around 22 ℃, with a humidity of 50 -60% and alternating night and day cycles for 12 h. Animal experiments were conducted after one week of feeding. All experimental procedures on animals were approved by the Ethics Committee of the hospital (approval number: SYXK (Su) 2018-0003) in accordance with the guidelines provided by the Committee for Purpose of Control and Supervision of Experiments on Animals (CPCSEA). The rats were anesthetized by intraperitoneal injection of 5% phenobarbital. A longitudinal incision of about 2 cm was made above the foramen magnum of the occipital bone while in a prone position. The subcutaneous tissue and neck muscles were bluntly separated, and the atlantooccipital membrane was exposed. The atlantooccipital membrane was then penetrated using the needle tip, resulting in the clear cerebrospinal fluid gushing out and fluctuating with breathing. A sterilized, closed-end PE-10 catheter filled with physiological saline was inserted 7 cm towards the tail end, with the catheter successfully reaching the spinal canal indicated by the sudden lateral sway or hind leg twitching of the rat tail. The catheter was secured, and the incision was sutured layer by layer to the back of the lumbosacral segment of the spinal cord. On the first day after surgery, all rats were randomly divided into 3 groups according to computer sequence numbers (n = 6), namely, the sham, the 10% lidocaine, the 10% lidocaine + DEX (25 µg/kg). For the 10% lidocaine group, an intrathecal injection of 10% lidocaine was performed using a volume of 20 µL (Zhang et al. 2022; Ding et al. 2021). Positive results for the lidocaine screening test were determined if the rats experienced paralysis of both lower limbs within 30 s or exhibited avoidance reactions when both upper limbs were clamped. Rats showing no reaction in both lower limbs and the disappearance of the tail clamp reflex were selected for the experiment. Rats with quadriplegia, mobility disorders, negative lidocaine test results, or unilateral limb paralysis after surgery were excluded. For the 10% lidocaine + DEX (25 µg/kg) group, rats were intraperitoneally injected with 25 µg/kg DEX in a 0.1 mL vehicle containing 2% DMSO (Sigma) and 0.9% NaCl (Meilunbio, China). The first injection was administered two hours after the successful establishment of the model, followed by daily injections until the eight day. The sham group received an equal volume of vehicle injections. The rats were killed on the eight day after injection in vivo.
Behavioral score
On days 0, 2, 4, 6, and 8, the motor recovery of different groups of rats was evaluated using Basso, Beattie, and Bresnahan (BBB) motor scores (Basso et al. 1995) by two independent examiners blinded to the grouping. The scores ranged from 0 to 21, with 0 indicating no motor function and 21 indicating normal performance. Rats were allowed to walk freely within a 90 cm2 field for 5 min, and the movement of their hind limbs was closely observed.
Nissl staining and hematoxylin-eosin (HE) staining
The paraffin sections were dewaxed and soaked in three changes of distilled water for 5 min each. They were then stained with methyl violet staining solution for 10 min. After washing the slide, it was differentiated in Nissl (Beyotime) solution for 5 s, followed by dehydration in a gradient alcohol solution. After removal in xylene, the sections were mounted on a glass slide using neutral adhesive. The changes in the number of Nissl bodies in the anterior horn of the spinal cord were observed using an optical microscope at the same location. For HE staining, the paraffin sections were stained with hematoxylin (Solarbio) aqueous solution for 5 min. The slides were rinsed three times with distilled water, differentiated in ammonia and acidic water for 30 s, and then soaked in distilled water for 15 min. Subsequently, the sections were stained with Eosin (Solarbio) for 2 min, followed by dehydration in gradient alcohol and xylene removal. After the slides were fixed with neutral gum, the tissue cavities and cell numbers of each group were observed under a microscope.
Flow cytometry for ROS measurement in tissues
A ROS assay kit was used to determine ROS levels according to the manufacturer’s specification (Beyotime). Spinal cord samples in each group were collected and homogenized under ice-cold conditions. Each sample was incubated in the dark for 20 min in 500 µL of DCFH-DA (10 µM). After replacing the loading buffer and eliminating residual DCFH-DA, sample measurements were immediately performed using flow cytometry (Biosciences), with excitation light at 488 nm and emission light at 525 nm.
Transmission electron microscopy (TEM)
As mentioned earlier, TEM observations were conducted on the ultrastructure of spinal cord samples from different groups. The spinal cord samples from each group were incubated overnight in 2% glutaraldehyde at 4℃, followed by fixation with 1% citric acid. After immersion in uranyl acetate, they were dehydrated with gradient acetone. Subsequently, the samples were embedded in epoxy resin and cut into 70–90 nm sections, followed by re-stained with lead citrate on a copper groove grid. The ultrastructure of mitochondria was observed using TEM (Chiyoda ku, Tokyo, Japan).
Statistical analysis
Graphpad version 9.0 (Graphpad Software Inc., San Diego, CA, USA) was used for data analysis and plotting. All data are represented as mean ± standard deviation. The Shapiro-Wilk test was used to evaluate the distribution of data. If there were three or more datasets, the comparison was performed using a one-way analysis of variance (ANOVA) with Tukey’s post-test. A P-value < 0.05 was statistically significant.

Comments (0)