
Remember me
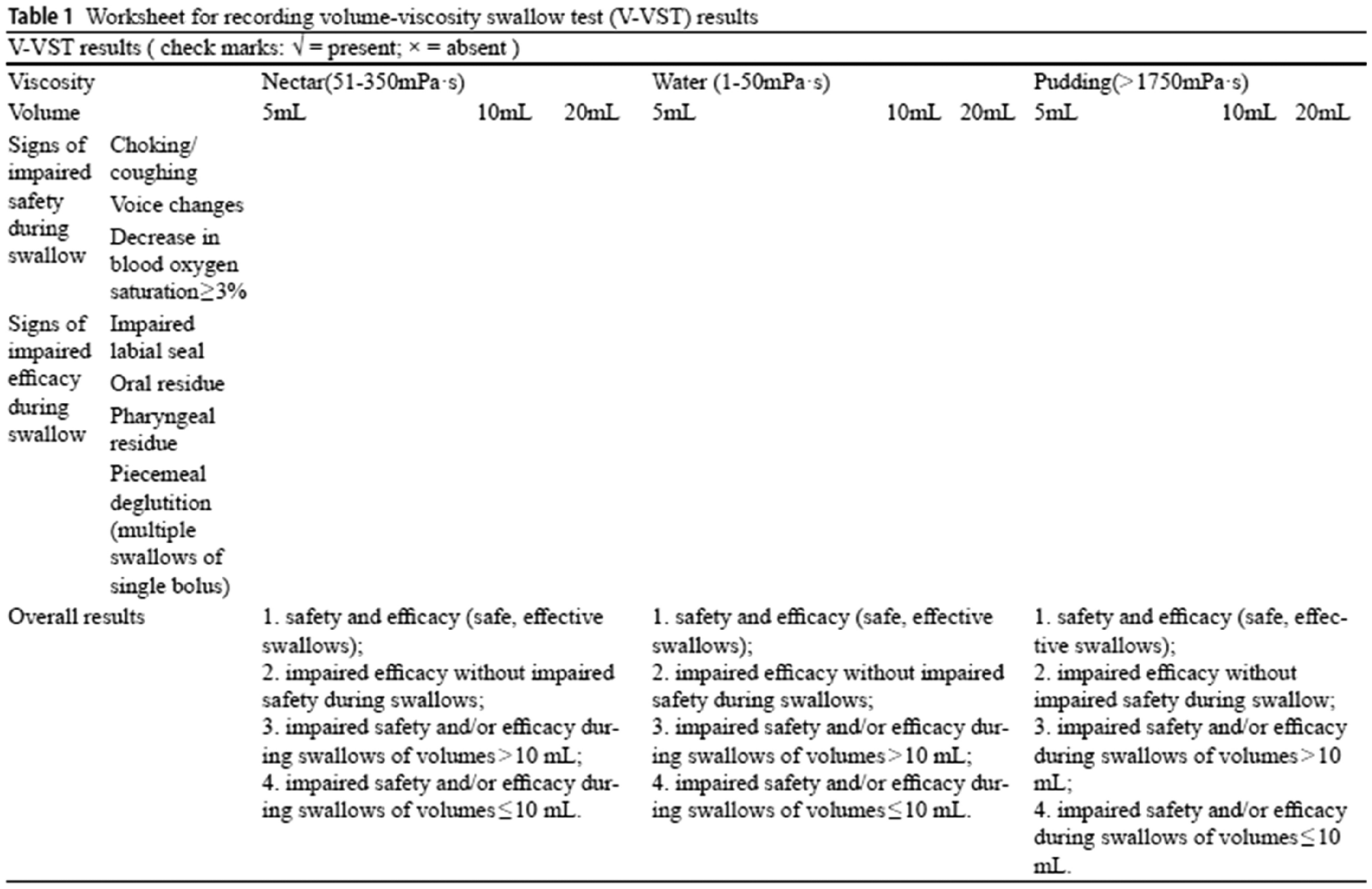





Thirty-nine patients, including 17 eCRSwNPs (11 males, 6 females), 9 non-CRSwNPs (8 males, 1 females) and 13 HCs (5 males, 8 females), were enrolled in the cohort. The demographic and clinical characteristics of the three groups are displayed in Table 1. Except for age and BMI, there was no difference among the three groups.
Table 1 Demographic characteristics of the cohortsDNA sequencing results of the gut microbiomeThe OTUs of the three groups were used to construct the petal diagram and phylogenetic tree (eFigure 1A, B). The dilution curve was gentle, indicating that the results obtained based on the sequencing depth could reflect the diversity of the gut microbiome in the samples (eFigure 2A, B).
At the phylum level, Firmicutes, Actinobacteria and Proteobacteria were the main phyla (Fig. 1A). Compared with those in the eCRSwNP group, the proportions of Actinobacteria in the non-CRSwNP (P = 0.0437) and HC (P = 0.0389) groups were significantly increased, while the proportions of Proteobacteria in the non-CRSwNP (P = 0.2584) and HC (P = 0.1837) groups were decreased. At the genus and species levels, the composition of the gut microbiome in the three groups is displayed (Figs. 1B, C). In summary, at different taxonomic levels, our results suggested the distinct distribution of the gut microbiome in the three groups.
Fig. 1
Comparison of relative taxa abundance among the HCs, eCRSwNP patients and noneCRSwNP patients groups at phylum, genus and species levels. A The bar chart of relative taxa abundance among the three groups at phylum levels. B The bar chart of relative taxa abundance among the three groups at genus levels. C The bar chart of relative taxa abundance among the three groups at species levels. n = 39, eCRSwNP patients = 11, noneCRSwNP patients = 9, and HCs = 13. eCRSwNP eosinophilic chronic rhinosinusitis with nasal polyps, HCs healthy controls
The diversity in the gut microbiome among groupsIn this cohort, there was no difference in intraindividual diversity among the eCRSwNP, noneCRSwNP and HC groups, as measured by the observed OTUs and Shannon and Faith’s phylogenetic diversity indices (eTable 1). Next, PCoA was used to assess the overall diversity of the gut microbiome (Fig. 2A, B). The gut microbiome of the eCRSwNPs, noneCRSwNPs and HC groups indicated partial but significant clustering in the PCoA diagram. Based on the weighted UniFrac distance index among the three groups (eTable 2), the significant distinctions in overall diversity between eCRSwNP and HC (P = 0.020) and between eCRSwNP and noneCRSwNP (P = 0.020) were significant, while those between noneCRSwNP and HC (P = 0.482) were not significant. Therefore, the gut microbial structure of the eCRSwNP group was significantly different from that of the noneCRSwNP and HC groups. Our results indicated that the gut microbiota may play a crucial role in the pathogenesis of eCRSwNP.
Fig. 2
Characteristics of gut microbiome composition in the HCs, eCRSwNP patients and noneCRSwNP patients. A Diagram of the LDA scores calculated at genus levels among HCs, eCRSwNP and noneCRSwNP groups. Only the LDA score > 2 are shown in the figure. B Diagram of the LDA scores calculated at genus level between eCRSwNP and HCs groups. C Diagram of the LDA scores calculated at genus level between noneCRSwNP and HCs groups. n = 39, eCRSwNP patients = 11, noneCRSwNP patients = 9, and HCs = 13. eCRSwNP eosinophilic chronic rhinosinusitis with nasal polyps, HCs healthy controls
Next, the gut microbiome composition of the three groups was clustered by PLS-DA (Fig. 2C). Our results indicated that the gut microbiome composition of the three groups was significantly distinct. The prediction models were established based on the nasal detected distinct genera of the three groups by using PLS-DA. The performance of the models achieved an AUC value of almost 1 (Fig. 2D). Our findings indicated the great potential of the gut microbiome as a noninvasive classifier for eCRSwNP diagnosis and may be recognised as a risk factor in the pathogenesis of eCRSwNP.
Differences in the gut microbiome structure among groupsThe DESeq2 method was performed to identify differences in the gut microbiome structure among groups (eTable 3). The relative abundances of 2 genera in the noneCRSwNP and HC groups were significantly different, and the relative abundances of 7 genera in the eCRSwNP and noneCRSwNP groups were significantly different. Compared with HCs, the abundances of Escherichia and Enterococcus were significantly reduced in the group at the genus level. Compared with those in noneCRSwNPs, the increased abundances in gut microbiota such as Enterobacter, Escherichia, Megamonas and SMB53 were observed in eCRSwNPs, and the abundances of Bifidobacterium, Streptococcus and Collinsella were significantly increased in noneCRSwNPs. These differential genera can be used to build a noninvasive classifier for the distinct abundant taxa between eCRSwNPs or non-CRSwNPs and HCs.
To identify the distinct abundant taxa among the eCRSwNP, noneCRSwNPs and HC groups, LEfSe analysis was performed on the gut microbiome composition. At the genus level, 14 bacterial taxa showed distinct relative abundances among the three groups (LDA score > 2.0, p < 0.05). Increased abundances of Clostridia, Clostridiales, Firmicutes, and Gemmiger were observed in the non-CRSwNP group, and increased abundances of Bifidobacterium, Actinobacteria, Bifidobacteriales, etc., were observed in the eCRSwNP group (Fig. 3A). Compared with HCs, it was found that the abundances of Turicibacter, Clostridium, Gemmiger, etc., were increased significantly in eCRSwNPs (Fig. 3B); the abundances of Peptostreptococcus, Eubacterium, Clostridium, etc., increased significantly in noneCRSwNPs (Fig. 3C).
Fig. 3
PCoA and PLS-DA analysis of the microbiome among the HCs, eCRSwNP patients and noneCRSwNP patients. A Bray–Curtis PCoA based on the relative abundance of OTU (99% similarity level). B Unweighted UniFrac PCoA based on the relative abundance of OTU (99% similarity level). C The PLS-DA analysis on OTUs among the HCs, eCRSwNP patients and noneCRSwNP groups. D ROC analysis for the predictive value of the predictive model constructed based on PLS-DA analysis. The AUCs of the HCs, eCRSwNP patients and noneCRSwNP groups almost are 1. n = 39, eCRSwNP patients = 11, noneCRSwNP patients = 9, and HCs = 13. AUC the area under the curve, eCRSwNP eosinophilic chronic rhinosinusitis with nasal polyps, HCs healthy controls, PCoA principal coordinate analysis, PLS-DA analysis partial least squares Discriminant Analysis
Correlation analysis between the gut microbiome and clinical variablesPartial Spearman’s rank-based correlation test performed on the age, sex, IgE, serum eosinophil count, serum eosinophil percent, BMI, SNOT-20, NO and LM scores was employed to explore the link between clinical variables and the disease-associated abundant taxa in all CRSwNP patients (Fig. 4A). At the genus level, the abundance of Haemophilus and Faecalibacterium positively correlated and Corynebacterium negatively correlated with LM CT scores. Dialister and Enterococcus were positively correlated, and Clostridium, Coprococcus and SMB53 were negatively correlated with SNOT-20. IgE and NO showed similar correlations with the gut microbiome. These results suggested that the faecal microbiota correlates with eCRSwNP disease severity. In addition, permutational multivariate analysis of variance results revealed that the above subject characteristics, such as age, sex and BMI, did not have a significant impact on the gut microbiota of different groups in our cohort (etable 4).
Fig. 4
The relationship between gut microbiome and clinical variables at the genus level. A Heat map for Spearman correlation analysis between gut microbiome and clinical variables at the genus level. B The patients with high abundance of Turicibacter (n = 13) had higher level of percentage tissue eosinophil compared to those with low abundance of the species (n = 13). C The patients with high abundance of Faecalibacterium (n = 13) had higher LM CT scores compared to those with low abundance of the species (n = 13). P < 0.05 is showed in the figure. *P < 0.05, **P < 0.01, ***P < 0.001. BMI body mass index, CRSwNP chronic rhinosinusitis with nasal polyps, eCRSwNP eosinophilic chronic rhinosinusitis with nasal polyps, EOS_NUMB absolute tissue eosinophil count, EOS_PERCENT percentage tissue eosinophil, HCs healthy controls, IgE immunoglobulin E, L_M_SCORE Lund-Mackay CT scores, sEOS_NUMB serum eosinophil count, sEOS_PERCENT serum eosinophil percent, SNOT20 sinonasal outcome test scores
In patients with CRSwNP, the degree of eosinophil infiltration and the intensity of the nasal mucosal inflammatory response played a crucial role in the prognosis and disease severity. At the genus level, the abundances of Escherichia and Turicibacter positively correlated with absolute tissue eosinophil count. Gemmiger and Turicibacter were positively correlated, and Lachnospiraceae Clostridium was negatively correlated with the percentage of tissue eosinophils. Similarly, Turicibacter was also found to be positively correlated with serum eosinophil count and serum eosinophil percentage. In addition, Parabacteroides negatively correlated with serum eosinophil percentage, while Parabacteroides and Oscillospira negatively correlated with serum eosinophil count.
To further clarify the relationship between the microbiome and clinical variables, the CRSwNP patients were divided into two groups based on the median levels of the gut microbiome. After adjusting for age, sex and BMI, multivariate logistic regression results revealed that CRSwNPs with a high abundance of Turicibacter were relevant to a higher percentage of tissue eosinophils (P = 0.032, OR = 1.052, 95% CI 1.004–1.102, Fig. 4B). A high abundance of Faecalibacterium was associated with higher LM CT scores (P = 0.047, OR = 1.272, 95% CI 1.003–1.613, Fig. 4C).
Comments (0)