
Remember me


A 15-month-old female (P1) born to non-consanguineous parents of Samoan origin (Fig. 1A, II.2) was noted in the first few months of life to have developmental delay and clinical dysmorphology including arthrogryposis. During the first 12 months of life, she had a significant history of severe and repeated respiratory and gastrointestinal illnesses. At 4 months of age, she had severe hypoxemic SARS-CoV2-induced pneumonia that required supplemental oxygen and was treated with high dose intravenous dexamethasone as per institutional guidelines. No passive SARS-CoV2 immunity was expected as maternal vaccination was commenced 2 months after the patient’s birth. At 6 months, she had severe enterovirus meningitis and gastroenteritis requiring prolonged hospital admission for fluid and feeding support; at 13 months she had severe adenovirus gastroenteritis requiring electrolyte and fluid therapy in hospital. Finally, 10 days after receiving MMR vaccination she became very unwell, presenting with protracted lethargy, irritability, intermittent fevers, rash, tachypnoea, anaemia, thrombocytopaenia, splenomegaly, hyperferritinaemia, hypofibrinogenemia and rapidly progressive hepatitis (Table 1). Clinical examination did not revealed any evidence of atopic disease, nor is there any evidence of eczema, or allergies to food or environmental allergens.
Fig. 1
IFNAR1 (c.1156G > T, p.(Glu386*) variant is loss-of function in T cells (A) Pedigree of a novel IFNAR1-deficient kindred. Familial segregation of the IFNAR1 c.1156G > T, p.E386X and DOCK8 c.3234 + 2T > C variants are shown. The affected individual P1 is represented by a black symbol (II.2). Individuals of unknown genotype are labelled “E?”. (B-E) T cell blasts were expanded from healthy donors (HD) and P1 by stimulating PBMCs with anti-CD2/CD3/CD28 mAbs. After 14 days, the cells were rested, and then stimulated in the absence or presence of (B, C) IFNb (type I IFN) or (D, E) IL-21. After 15 min, cells were harvested, fixed and permeabilised and then stained with mAb specific for phospho-STAT1. FACS plot (B, C) are representative of pSTAT1 induction in T-cell blasts from HD or P1. The data in (C) and (E) represent the fold-change in pSTAT1 gMFI in T-cell blasts stimulated with IFNβ or IL-21, respectively, relative to unstimulated cells. Each point represents data from an independent experiment
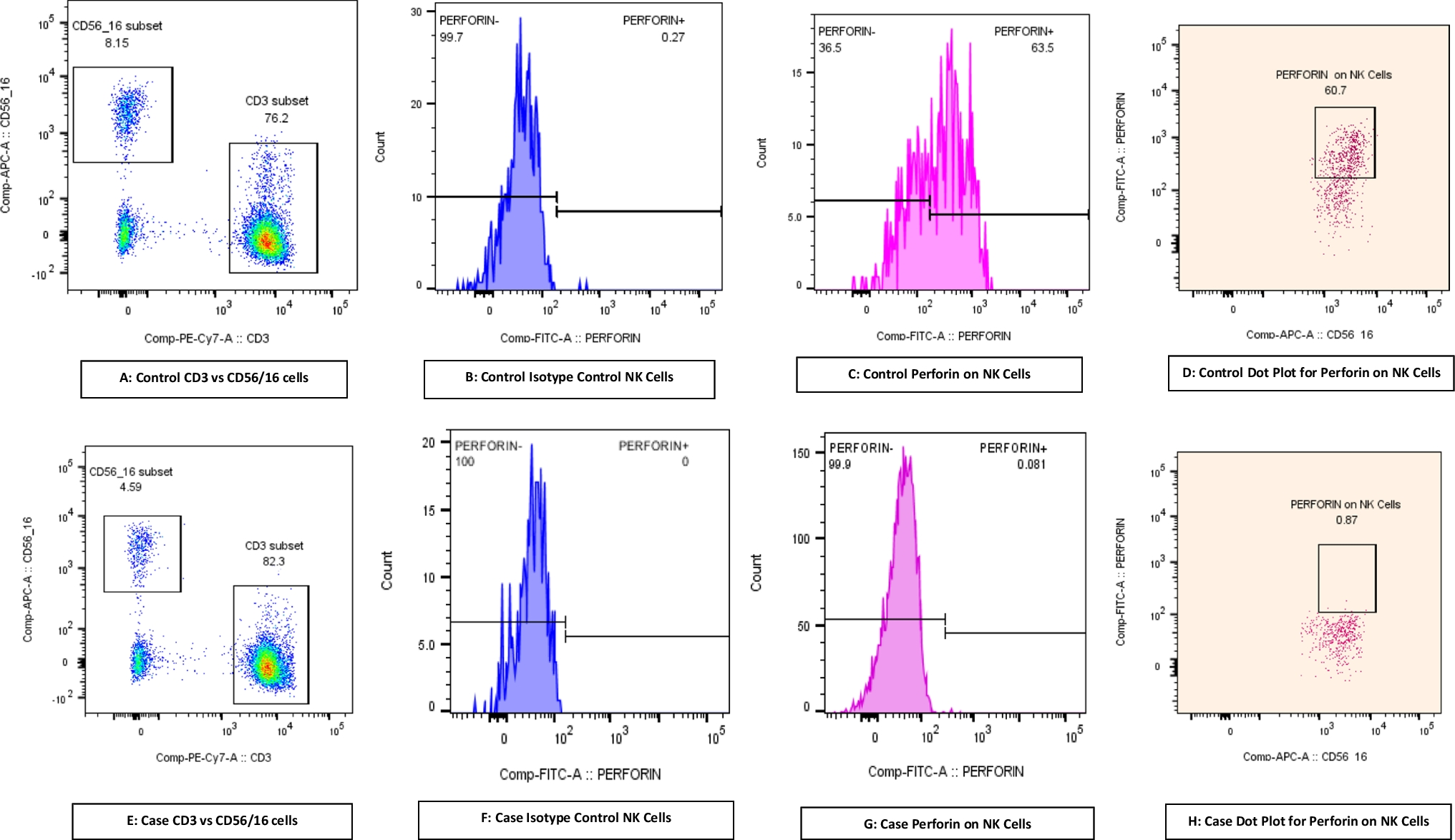
Prompt immunological evaluation to investigate possible primary hemophagocytic lymphohistiocytosis (HLH) was undertaken. NK cell degranulation (CD107a expression), and assessment of intracellular perforin, XIAP, SAP and surface HLA-DR expression by flow cytometric assays were within laboratory reference ranges of healthy donors. While soluble CD25 and ferritin levels were elevated, fibrinogen levels were reduced (Table 1) and a bone marrow aspirate did not show evidence of hemophagocytosis. Further immune work-up revealed panlymphopenia and hypergammaglobulinemia (Table 1). Overall, P1 developed a hyperinflammatory state that included some features of HLH. Interestingly, HLH was recently described in an unrelated case of IFNAR1 deficiency [2].
Table 1 Cellular, serological, inflammatory and infectious features of P1Extensive metabolic and malignant screens were also negative, while MRI brain, and chest/abdomen/pelvis CTs were normal. Furthermore, an extended infectious screen confirmed measles vaccine strain in the urine, and mumps and rubella viruses – but not measles - in nasopharyngeal and respiratory secretions. To exclude neuroinflammation and/or infection, CSF analysis was performed. Cytology and biochemistry were normal. Extensive CSF cultures and viral PCRs were negative, including for adenovirus, CMV, enterovirus, HSV1/2, parechovirus, JCV, WT and vaccine measles). Measles IgG was also non-reactive in CSF (Table 1). The patient has made a full recovery (current age: 30 months) and remains on Ig replacement therapy and regular surveillance. P1 has 2 healthy siblings, currently aged 4 yrs (II.1) and 4 months (II.3, Fig. 1A).
Outcomes of Genetic TestingBased on clinical phenotype of LAV-associated disease with hyperinflammatory complications, and the ethnicity of the patient and parents, IFNAR1 deficiency was suspected. Urgent whole genome sequencing was performed on whole blood DNA from the patient, with initial analysis focussed on genes known to cause metabolic, immunological, aortopathy and connective tissue disorders, as well as congenital hypothyroidism, and arthrogryposis. This identified three clinically relevant variants in P1:
[1] a pathogenic homozygous nonsense variant in IFNAR1 (c.1156G > T, p.Glu386* LOF) as previously described in patients from the Pacific Islands [15];
[2] a likely pathogenic heterozygous variant in FBN2 (c.4346-1G > C, p.?), and.
[3] a homozygous variant of uncertain significance (VUS) in DOCK8 (NM_203447.4, c. 3234 + 2T > C) predicted to abolish the highly conserved + 2 donor splice site (phyloP score 7.85 [-19.0-10.9]), phastCons score: 1 [0, 1]; http://compgen.cshl.edu/phast/) at the intron 26/exon 26 junction (Fig. 1A). As expected, genotyping each parent (I.1, I.2) revealed them to be heterozygous for the variants in IFNAR1 (c.1156G > T, p.Glu386* LOF) and DOCK8 (c. 3234 + 2T > C) (Fig. 1A).
Heterozygous pathogenic variants in FBN2 have been associated with congenital contractual arachnodactyly [23]. The FBN2 variant idenitifed in P1 was absent from gnomAD and has not been previously reported. It was considered to be pathogenic as parental segregation revealed it to be de novo in P1; this private FBN2 variant confirmed a diagnosis of congenital contractual arachnodactyly in P1. Thus, the IFNAR1 and DOCK8 variants were therefore investigated in more detail.
The IFNAR1 Glu386*Variant is LOFThe IFNAR1 (c.1156G > T, p.(Glu386*) variant identified in P1 has previously been shown to be LOF in over-expression systems and functional analysis in fibroblasts generated from an affected individual [15]. As the functional requirements of type I IFN signalling in host defence can differ between immune and non-immune cells [3, 7, 13], it was important to establish the IFNAR1 Q386* variant was also deleterious in leukocytes. To test this, T cell blasts were generated from PBMCs from P1 as well as healthy donors (HD), and then assessed for induction of STAT1 phosphorylation in response to IFNβ. In the presence of IFNβ, STAT1 phosphorylation was increased 5-10-fold in T-cell blasts from HD compared to unstimulated cells (Fig. 1B left panel, Fig. 1C). In contrast, no pSTAT1 could be detected in IFNβ-stimulated T-cell blasts from P1 (Fig. 1B right panel, Fig. 1C). Importantly, induction of pSTAT1 in T-cell blasts from HD and P1 stimulated with IL-21 was intact (Fig. 1D, E), establishing that the defect in response to IFNβ in P1 T cells was specific and not a result of generalised impairment of cytokine signalling. Thus, similar to our previously reported patients of Western Polynesian ancestry [15], IFNAR1 was LOF in P1.
DOCK8 3234 + 2T > C – In Silico Characteristics and Population GeneticsThe homozygous DOCK8 variant (NM_203447.4, c. 3234 + 2 T > C) in P1 is localized in intron 26, 2 bp after the exon 26 splicing donor site (Fig. 2A). The T > C substitution is reported to be likely pathogenic in dbSNP database (https://www.ncbi.nlm.nih.gov/snp/, rs756871628) and ClinVar (https://preview.ncbi.nlm.nih.gov/clinvar/, 573664). In silico analysis using multiple programs (MaxEntScan, NNSplice, GeneSplicer) predicted the variant will have an adverse effect on the splicing donor site, potentially leading to abolition of splicing and skipping of exon 26 (Fig. 2A). However, SpliceSiteFinder-like program predicted the variant would only decrease the donor site usage by 7.5% (WT score: 95.6%; variant score: 88.5%) and thus have only a modest impact on splicing. The variant has a combined annotation-dependent depletion (CADD) score of 32, well above the mutation significance cutoff (MSC) for DOCK8 (13.3) (Fig. 2B) [24, 25].
Fig. 2
DOCK8 expression is intact in lymphocytes from the IFNAR1-deficient patient harbouring the DOCK8 c.3234 + 2T > C variant (A) schematic of DOCK8 genomic sequence representing the 5’ region of exon 26 (blue), the 3’ region of intron 26 (black) and the variant found in P1 (red). Prediction of the cDNA sequence is reported for both WT DOCK8 and DOCK8 c.3234 + 2T > C. (B) Minor allele frequency (MAF, x-axis) and CADD scores (y-axis) for missense (green circles) and essential splicing (purple circles) variants in DOCK8 reported as homozygous in the public database gnomAD (GRCh37). The DOCK8 c.3234 + 2T > C variant found in P1 (indigo circles) is the only homozygous essential splicing variant previously reported in gnomAD. The mutation significance cut-off (MSC, 95% confidence interval, y axis) is represented by the dotted line. (C, D) PBMCs from healthy donors (HD), P1 and a confirmed DOCK8-deficient patient were fixed and permeabilised and stained with anti-DOCK8 mAb. Intracellular expression of DOCK8 was then determined. (A) DOCK8 expression (blue histogram) in PBMCs from a HD (left) and IFNAR1-deficient patient P1 (right) relative to staining with isotype IgG control mAb. (B) Overlay of DOCK8 expression in PBMCs from HDs (#1-#4), P1 (left) or a DOCK8-deficient patient (right). (E, F) exons 25–27 of DOCK8 were PCR amplified from PBMCs of healthy donors and P1. (E) agarose gel depicting amplified products, including 2 products for P1. (F) Sequencing of amplified PCR products from HD (left) and P1 (right) showing the higher molecular weight band from P1 corresponds to WT DOCK8 cDNA, while the lower band lacks exon 26. (G, H) whole cell lysates were prepared from T blasts expanded from healthy donors (HD), P1 and a confirmed DOCK8-deficient patient. DOCK8 expression was assessed by SDS-PAGE and Western blotting. Detection of GADPH was used as a loading control. (G) representative of western blot from 3 different experiments. (H) summary data depicting expression of DOCK8 protein in lysates from 3 unrelated healthy donors and P1, relative to GAPDH
Interestingly, the variant appears common in certain populations. Specifically, 28 heterozygous individual and one homozygous individual are present in gnomAD (v4.0.0), with 25/29 individuals being either Admixed American (n = 10), or ‘remaining’ (n = 15) ethnicity. There were no homozygotes for any other predicted LOF variants in DOCK8 in any public database. The DOCK8 c.3234 + 2T > C variant was not found in 2844 healthy elderly adults from the Medical Genome Reference Bank (MGRB) [26]. The ethnicity of the majority of the MGRB is non-Finnish European, with no known Polynesian representation. In terms of available population frequencies, the DOCK8 c.3234 + 2T > C variant has a MAF of 0.108 in Vanuatu in the Western part of Western Polynesia [27], but there is a lack of published data for other populations. In terms of variant distribution however, retrospective analysis of genomic data from > 25 000 patients who had undergone gene panel/WES/WGS for the diagnosis of various conditions found this variant in a heterozygous state in one individual from the Philippines, as well as 12 individuals of Polynesian ancestry (most residing in Australia or Aotearoa New Zealand) including one of the previously-reported patients with the homozygous IFNAR1 c.1156G > T; pGlu386* variant [P3 in reference [15]]. The occurrence in ‘Admixed Americans’ on the gnomAD database may reflect a broader distribution outside of Polynesia, or could potentially reflect ethnicity from, for instance, American Samoa or Hawaii in North Western Polynesia. Thus, similar to the IFNAR1 c.1156G > T variant, the DOCK8 c.3234 + 2T > C variant appears to have a high MAF in Polynesians and ‘Admixed Americans’.
Effect of the Homozygous DOCK8 Variant on Protein ExpressionWhile the clinical phenotype of P1 was not strictly suggestive of DOCK8 deficiency, she was only 15 months old and clinical onset of DOCK8 deficiency can be variable within the first 5 years of life [28,29,30]. Furthermore, due to a paucity of reports of DOCK8-deficiency in the Pacific regions, it is possible the clinical phenotype of affected individuals in this geographical area may differ from that reported for DOCK8-deficiency in the Americas, Europe and the Middle East [18, 29, 31, 32]. Lastly, as the DOCK8 variant was homozygous and located in a splice region it was important to assess potential pathogenicity to determine if P1 also had DOCK8 deficiency, as the treatments for IFNAR1-deficiency and DOCK8-deficiency are different [3, 28,29,30].
To investigate the impact of the DOCK8 c.3234 + 2T > C variant, we first assessed intracellular expression by flow cytometry [18,19,20,21]. Similar levels of DOCK8 were detected in PBMCs from HDs and P1 (Fig. 2C left panel, Fig. 2D). This contrasted a patient diagnosed with DOCK8 deficiency (homozygous c.2142G > A variant, p.W714*), whose leukocytes clearly lacked DOCK8 protein (Fig. 2D [right panel]).
The DOCK8 c.3234 + 2T > C variant was predicted to cause skipping of exon 26 of DOCK8, thus resulting in expression of a truncated protein (Fig. 2A). It was possible that this shorter protein could still be detected by the mAb used for flow cytometry. To explore this further, we performed PCR using primers that would amplify across exons 23–29. This amplified a 798 bp product from cDNA generated from PBMCs of 3 HDs (HD1-3; Fig. 2E). Interestingly, 2 products were amplified from P1 (Fig. 2E): one corresponding to that detected in HDs, and a shorter product lacking exon 26 (114 bp encoding 38 amino acids; Fig. 2E, F).
As this shorter cDNA represented a relatively low proportion of amplified product, it was unlikely to be the dominant protein isoform. Indeed, Western blotting using a DOCK8-specific Ab detected a protein of comparable molecular weight in lysates extracted from T-cell blasts expanded from 3 HDs and P1 (Fig. 2G, H). The absence of detectable DOCK8 protein in lysates from T cells expanded from a known DOCK8-deficient patient confirmed the specificity of the anti-DOCK8 mAb used (Fig. 2G). Thus, consistent with the flow cytometry data, and despite the presence of a truncated cDNA transcript, the DOCK8 variant detected in P1 does not appear to contribute substantially to expression of total DOCK8 protein.
T Cells from P1 lack Phenotypic Signatures Features of DOCK8-Deficient T CellsTo provide further evidence that the DOCK8 c.3234 + 2T > C variant was benign, we assessed the phenotype and function of T cells from P1 for evidence of aberrant DOCK8 function once the acute hyperinflammatory process had resolved. Our previous studies identified a suite of detectable pathognomonic cellular defects in DOCK8-deficient patients, even prior to disease onset [18,19,20,21,22]. Typical DOCK8-deficient patients have an inverted CD4:CD8 ratio (~ 1:2 vs. 2–3:1 for HD), and aberrant CD4+ and CD8+ T cell compartments (reduced naïve cells/increased TEM and TEMRA cells) compared to HDs [18, 20, 21] (Fig. 3A-C, upper and middle panels; Fig. 3D, E). Similarly, the senescent/exhaustion marker CD57 is expressed on increased proportions of DOCK8-deficient CD8+ T cell subsets compared to corresponding subsets in HD [18, 19, 22] (Fig. 3F). When these parameters were determined for P1, we also found a decreased CD4:CD8 ratio (1:2.5, Fig. 3A) which probably reflects hyper-active immunity during convalenscence post-MMR vaccination. Despite this, proportions of naïve and memory CD4+ T cell populations, as well as expression of CD57 on CD8+ T cell subsets, were comparable to HDs, as well as other patients with the homozygous IFNAR1 (c.1156G > T, p.(Glu386*) LOF variant (Figure Fig. 3B-F). Furthermore, CD8+ T cells in P1 were predominantly naïve, which is in stark contrast to DOCK8-deficiency, even for age-matched patients (Fig. 3B-E).
Fig. 3
Comparison of the phenotype and function of T cells from patients with IFNAR1-deficiency versus DOCK8-deficiency (A-F) PBMCs from HDs, one DOCK8 deficient patient, 2 IFNAR1-deficient patients or P1 were stained with mAbs against CD3, CD4, CD8, CD45RA, CCR7 and CD57. (A-C) representative FACS plots and (D-E) summary graphs depicting proportions of: (A, D) CD4+ and CD8+ T cells within CD3+ T cells, (B-E) naïve, TCM, TEM and TEMRA subsets within CD4+ (B, D) and CD8+ (C, E) T cells in HDs, and the indicated patients. (F) expression of CD57 on CD8+ naïve, TCM, TEM and TEMRA subsets, and the indicated patients. (G, H) CD45RA− memory CD4+ T cells were sorted from healthy donors (HD), a confirmed DOCK8-deficient patient, and P1 and then stimulated in vitro for 5 d with anti-CD2/CD3/CD28 mAb beads. After this time, secretion of (G) Th2 cytokines IL-4, IL-5 and IL-13 and (H) Th17 cytokines IL-17 A, IL-17 F and IL-22 was determined by cytometric bead array
Intact Cytokine Production of T Cells from P1Another characteristic of DOCK8 deficiency is aberrant cytokine production by memory CD4+ T cells. Specifically, Th2 cytokines IL-4, IL-5 and IL-13 are dramatically increased in DOCK8-deficient memory CD4+ T cells, while that of Th17 cytokines (IL-17A, IL-17F and IL-22) is decreased compared to memory CD4+ T cells from HD [18,19,20,21]. The cytokine profile was tested by isolating memory CD4+ T cells from HDs, a known DOCK8-deficient patient, and P1 and measuring secretion of these cytokines after 5 days of in vitro stimulation. As expected, production of Th2 cytokines was exaggerated for memory CD4+ T cells from a confirmed DOCK8-deficient patient compared to HDs, while production of Th17 cytokines from DOCK8-deficient memory CD4+ T cells was severely impaired (Fig. 3G, H). Importantly, Th2 cytokine production by P1’s memory CD4+ T cells was less than HDs, while Th17 cytokines were intact, greatly exceeding levels produced by DOCK8-deficient memory CD4+ T cells (Fig. 3G, H). Combined, our molecular, cellular, and functional studies establish that the DOCK8 c.3234 + 2T > C variant is benign and highly unlikely to be pathogenic in our patient.
Comments (0)