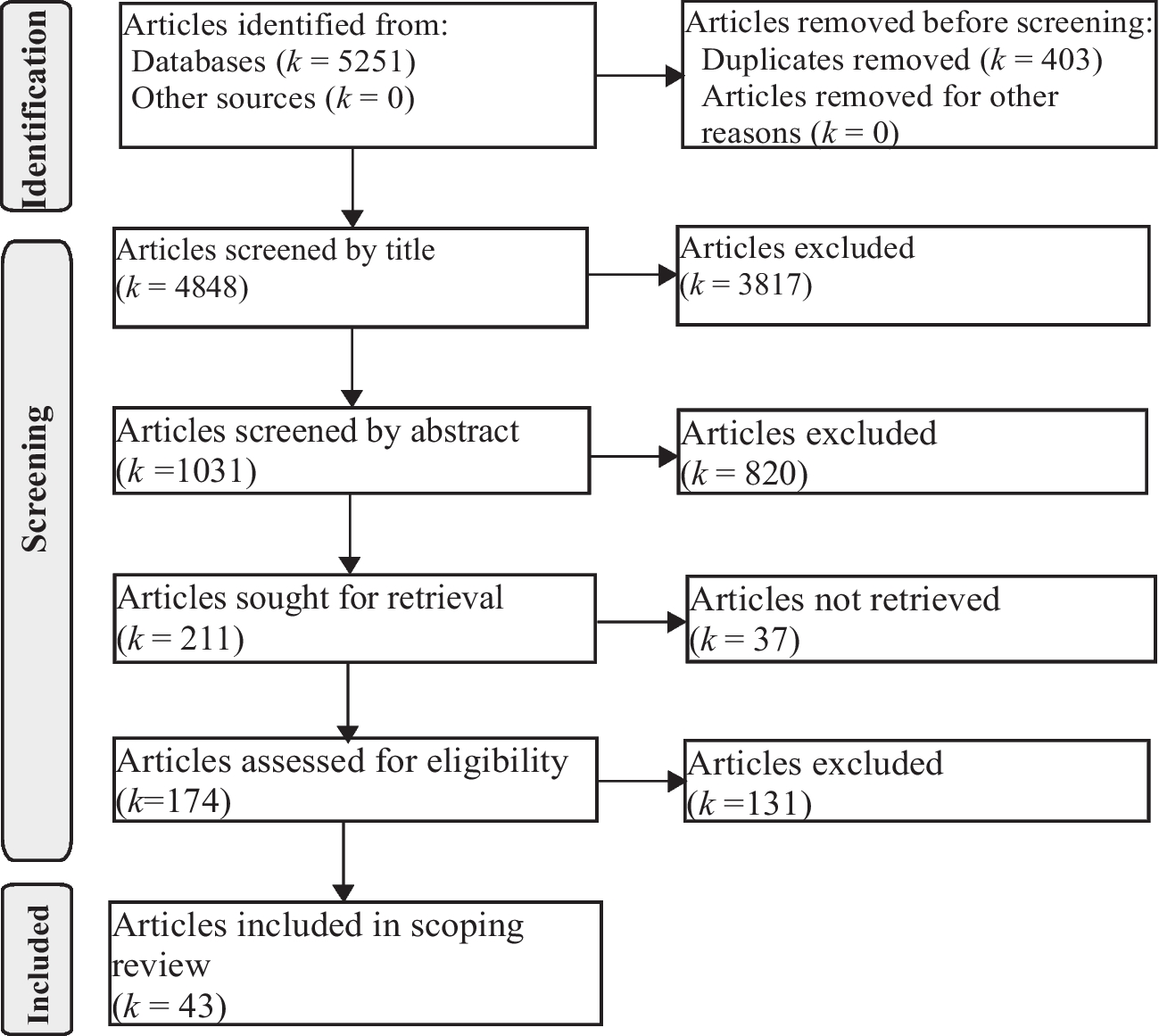
ACH is the leading cause of disproportionate short stature, and individuals with ACH encounter various medical, functional, and psychosocial challenges over their lifetimes. Early intervention and regular follow-up are essential for effectively managing potential complications in individuals with ACH. This study aimed to identify the most common clinical features and associated morbidities among 68 patients with ACH diagnosed in our clinic over the past 25 years.
Short stature is one of the main concerns in ACH. Moderate to marked short stature is present in all affected individuals. Obesity is another concern in ACH [18]. However, excessive weight gain usually becomes noticeable during early childhood, and in adulthood, obesity can exacerbate morbidity associated with lumbar stenosis, cause generalized joint issues, and contribute to the onset of cardiovascular complications [12]. Childhood obesity rates in ACH have been estimated to range from 0 to 10%, which is significantly lower than the rates observed in adults. Among adolescents, the prevalence of overweight and obesity has been reported to be as high as 56% [19, 20]. In our study, we evaluated the body mass index of 37 patients with ACH during the most recent follow-up. In the age group of 0–16 years, only two patients were obese according to the standards of ACH. However, three patients for whom BMI data were available in adulthood, one was overweight and two were obese. Although this suggests that the prevalence of adult obesity is increasing, the number of adult patients was insufficient for a clear interpretation.
Individuals with ACH generally have normal cognitive development and function [21]. However, when compared with controls, they have specific differences in development, including delayed and unusual motor development and language-related problems [22]. Mild to moderate hypotonia is typical during infancy, making it difficult for infants to support their heads. This, along with variations in body habitus, contributes to motor development delays [23,24,25]. Except in cases of hydrocephalus or other central nervous system problems, intelligence is normal [26]. Nevertheless, a small minority of children with ACH will be more seriously delayed, demonstrate significant learning disabilities, and may have autism spectrum disorders and/ or a cognitive disability [22]. Although the frequency of such problems has not yet been well documented, it is suggested that it accounts no greater than 10% [22]. In this study, the majority of patients initially had a delay in gross motor skills during infancy, as noted in informal assessments. However, they eventually caught up with their peers. Formal evaluations, using either the Denver II Developmental Screening Test or WISC-R, indicated that four patients had borderline intelligence, one had mild intellectual disability, one had moderate intellectual disability, and two (a twin pair) had learning disabilities and attention deficit hyperactivity disorder (ADHD). Notably, two additional patients (n = 2/68, 2.9%) were also found to have serious delays. The brain MRIs of these patients showed foramen magnum stenosis with hydrocephalus in four patients, cervical stenosis in two patients, cervical stenosis and cerebral atrophy in one patient, and thin corpus collosum in one patient.
Symptomatic hydrocephalus is rare in ACH [7, 27, 28]. However, around 5% of children with ACH may develop symptomatic increased intracranial pressure requiring intervention [29]. The reported percentage of individuals requiring treatment with a VP shunt or a subdural tap ranged between 2–5% [30,31,32]. In the present study, about one third (n = 13/47, 27.7%) of patients had hydrocephalus. However, VP shunt surgery was required in only one (7.7%) patient.
Population-based studies indicate that without evaluation and treatment, the excess risk of death for infants with ACH could reach 7.5% in the first year of life due to issues with the craniocervical junction [11]. This risk seems to be related to central apnea caused by damage to the respiratory control centers. This risk could be reduced to as little as 0.3% with evaluation and neurosurgical treatment [33]. In the present study, following a neurological evaluation and cranial MRI, a total of five patients (n = 5/47, 10.6%) with ACH underwent intervention.
Foramen magnum stenosis is a well-recognized, serious, and potentially life-threatening complication in ACH. It might clinically present with sleep-disordered breathing, hypotonia, or hypertonia with increased reflexes and extensor plantar responses; however, children and infants with foramen magnum stenosis can also be asymptomatic [7]. To ensure optimal monitoring of this potentially life-threatening complication, guiding principles have been developed [34]. These include routine clinical monitoring of infants and young children, scheduled magnetic resonance imaging screenings, referral of suspected cases to a neurosurgeon, combined assessments to inform decompression decisions, collaborative decision-making on proceeding with decompression, and management strategies for older children with previously undetected foramen magnum stenosis [34]. In the present study, foramen magnum stenosis was detected in more than half (53.2%) of the patients, yet six of them (24%) had headache, nine had apnea (36%), and three (12%) required surgical intervention. Risks for apnea-related death, as well as high cervical myelopathy and paralysis, result from the foramen magnum's growth being out of phase with that of the spinal cord. High cervical myelopathy can also be caused by compression of the cervicomedullary cord, typically manifesting in young children as disproportionate and persistent hypotonia, weakness, asymmetric reflexes, and hyperreflexia [35]. Therefore, from the time of diagnosis, every patient should undergo a thorough neurological examination, neuroimaging, and PSG evaluation. Symptomatic spinal stenosis, affecting L1-L4, is the most prevalent medical condition in adults [36]. In our ACH group, headache was noted in six patients; arm pain, weakness, and paresthesia in one; and leg pain, weakness, and paresthesia in two patients. These symptoms frequently manifested in late adolescence and adulthood.
Thoracolumbar junction kyphosis affects 89% of infants with ACH, but it usually resolves spontaneously over time [37]. The prevalence of kyphosis is lower, ranging from 19 to 35%, in children of walking age (over 3 years) and adolescents [38]. In our study, six patients (11.5%) exhibited kyphosis at ages over 3 years. Scoliosis is another common skeletal finding, affecting 60% of patients at an average age of 18 years [39]. In the present study, scoliosis was the most common musculoskeletal finding, observed in 28.8% of patients, yet none of the patients required surgical intervention. Only three patients had been using a corset for scoliosis. Scoliosis in patients was determined by considering both clinical and radiographic findings, however, the lower incidence of scoliosis among our patients was thought to be due to their younger average age (median: 6 years; range: 0–28 years) and missing follow-up data.
Otitis media and hearing loss are common problems in ACH [22]. Otitis media affects about 80% of all children with ACH at some point in their lives. In the present study, more than half (54.7%) of the patients had otitis media, and 9.4% experienced hearing loss. Previous studies reported higher rates of otitis media (80%) and hearing loss (37%) in patients with ACH than in our patients, which was thought to be related to missing data of some patients not continuing their regular follow-up after diagnosis [40]. Of patients with otitis media, about one-third (30.2%) had a history of ventilation tube placement. Snoring, mouth breathing, sleep apnea, and obstructive sleep apnea are common breathing disorders in ACH. Among our patients, snoring was a very common symptom (94.3%). In their clinical histories, apnea accompanying snoring was observed in 16 patients (30.2%). Obstructive sleep apnea (OSA) was detected in 14 of 20 patients for whom PSG evaluation data were available. PSG should be performed on all patients with ACH following diagnosis, since clinical history is a poor predictor of apnea [41]. Of note, despite the absence of symptoms such as snoring or apnea, OSA was detected with PSG in two of our patients.
Approximately half of patients (n = 31; 45.6%) have undergone at a minimum one surgery related to ACH such as ventilation tube insertion, adenoidectomy, neurosurgery, ortopedic surgery. This rate was 80% in the United States, 75.7% in the Japanese, and 72% Europe ACH cohort [42,43,44]. It has been suggested that inadequate patient data may be the reason for the lower rate of surgery in this study.
Middle ear procedures were the most prevalent type of surgeries as in the previous studies [42,43,44]. Lengthening surgery was performed in six patients (11.5%) in our cohort. The rate of lengthening surgery in patients with ACH varies considerably among countries, ranging from 1.2% in the United States to ~ 60% in Japan and up to 90% in ~ Spain [42]. Besides medical approaches, societal perspectives on disease and cultural values are likely to play a role in the development of such serious differences. For the same considerations, the rate of growth hormone use in treatment in a study in Japan was 75.7%, whereas in this study, three patients (4.4%) received growth hormone therapy [44].
Interestingly, a few unusual findings in some of our patients caught our attention. In one of the twin pairs, headache, learning difficulties, and ADHD were present and a brain MRI displayed foramen magnum stenosis and craniocervical junction compression. The fundus examination revealed papilledema, but neurological examination was completely normal, and an increase in intracranial pressure was not evident. Nevertheless, after receiving medical treatment with acetazolamide, the papilledema improved. Patients with ACH with papilledema have previously been reported; however, unlike our patients in discussion, hydrocephalus and high intracranial pressure were generally present in these patients [45].
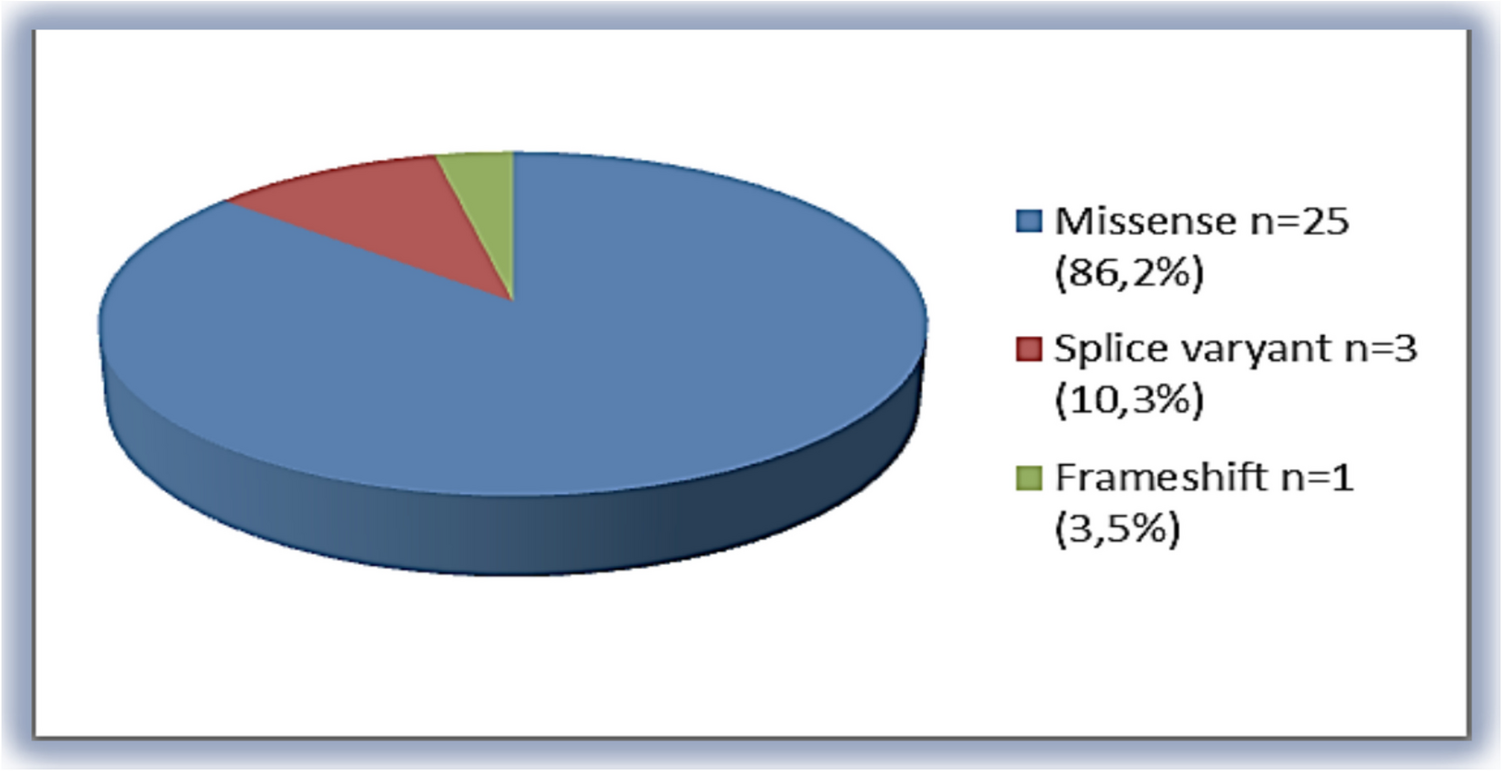
The other interesting finding was a patient with hypergonadotropic hypogonadism and streak gonads with a karyotype of 46,XX. To the best of our knowledge, gonadal dysgenesis has not previously been reported in ACH. Our patient exhibited hypogammaglobulinemia and severe intellectual disability as well. Due to atypical findings, advanced molecular analysis including chromosomal microarray analysis and exome sequencing was performed in addition to FGFR3 sequence analysis. Apart from the FGFR3 c.1138G > A heterozygous variant, no other pathogenic or clinically significant variant was identified through these tests.
Lastly, during follow-up, one of the patients, who showed both phenotypic and radiological findings consistent with ACH, displayed acanthosis nigricans and global developmental delay which suggested a diagnosis of severe ACH with developmental delay and acanthosis nigricans (SADDAN; MIM#616482). Physical examination revealed diffuse hyperpigmentation on the neck and trunk with dryness of the skin. There was no evidence of insulin resistance or adrenal insufficiency. Therefore, the most common variant in SADDAN c.1949A > T; p.Lys650Met was initially checked which was normal. A heterozygous FGFR3 pathogenic variant, c.1138G > A was detected in this patient.
Acanthosis nigricans in patients with ACH has rarely been reported in the literature; however, a recent study showed that it is present in 10% of patients. It is more likely to occur in the non-white population and has typically been observed to first appear in prepubertal or adolescent years [46]. In our patient group, only one patient (1.5%) had acanthosis nigricans, which may be related to the patients' ages and the fact that some of the patients admitted were from the non-white population. At the last examination, 54 patients (79.4%) were preadolescents (under 10 years old), and we plan to continue monitoring these patients for acanthosis nigricans.
In previous studies, mortality in children under 4 years of age was often sudden death with acute brainstem compression [12, 33]. The five patients (7.3%) who deceased in our study were also due to pneumonia. One of them had Down syndrome and died due to an aspiration pneumonia and the other one died due to a postoperative pneumonia. Although recurrent pneumonia and serious infections are an expected finding in Down syndrome, life-threatening pneumonia in patients with ACH is not as common as it was in our study. These patients had symptoms such as severe sleep apnea and difficulty swallowing, so we thought they might have recurrent infections associated with complications secondary to foramen magnum stenosis. However, we did not have data to support this because two of our five patients who died did not have a brain MRI before death and the other three did not have foramen magnum stenosis.
This study has several limitations, one of which is the missing follow-up data for some of our patients. Although this study includes patients of various ages, we were unable to plot the distribution of height SD scores due to missing data. Secondly, formal assessments for clinical findings, including intellectual disability and apnea, were not available for some patients. Thirdly, information on multidisciplinary and adulthood follow-up is missing.
In conclusion, ACH is commonly viewed as a mild condition, yet its associated health risks and potential for mortality are often underestimated. Early and continuous monitoring for serious complications from diagnosis onwards is essential to prevent early death, handle future health issues, and enhance the overall quality of life. Despite being the most prevalent form of non-lethal skeletal dysplasia, ACH's complications are not limited to orthopedic problems alone. This highlights the need for a comprehensive approach by a multidisciplinary healthcare team to cater to the varied needs of individuals with ACH.

Comments (0)