Screening of genes related to ferritinophagy-mediated ferroptosis in sepsis
The GSE165226, GSE179554-6 h and GSE179554-12 h data sets related to mouse sepsis model were selected, and the common deg intersecting genes in these three data sets were screened according to log2FC > 1.4, and the genes involved in sepsis ferritinophagy-mediated ferroptosis were identified.
Cell culture
Mouse lung epithelial cells MLE-12 (CRL-2110™, ATCC, Manassas, VA, USA) were grown in a specialized medium (CM-0680, Procell, Wuhan, China) containing various components required for its growth. Incubation of the culture was set at 37 °C in a suitable incubator with 5% CO2 in air atmosphere. To build sepsis models in vitro, LPS (1 mg/mL, Sigma, St. Louis, Missouri, USA) was used to stimulate MLE-12 cells for 24 h.
Small interference RNAs (siRNAs), plasmid construction, and transfection
For knocking down Mir22hg in vitro, Mir22hg siRNA (si-Mir22hg) (RiboBio, Guangzhou, China) was transfected into cells with Lipo3000 (Invitrogen, California, USA). Non-targeting siRNA (siNC) (RiboBio) was used as a negative control. YTHDC1 and Angptl4 full-length sequences were PCR-amplified with the primers. The PCR products were cloned into the pcDNA3.1(+) (Invitrogen) to generate YTHDC1 and Angptl4 overexpression plasmids (YTHDC1-OE and Angptl4-OE). For cell transfection, the Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) was applied following the instructions for usage provided by the manufacturer.
Determination of lipid reactive oxygen species (ROS)
BODIPY 581/591 C11 reagent (Invitrogen, California, USA) was used to measure the lipid ROS level. Briefly, after the indicated treatment, cells were dyed with a 5-µM reagent and incubated for 30 min (Zhang et al. 2022a, b, c). Intracellular fluorescence intensity was measured using a flow cytometer (BD Accuri C6 Plus, BD Biosciences, USA).
Cell viability analysis
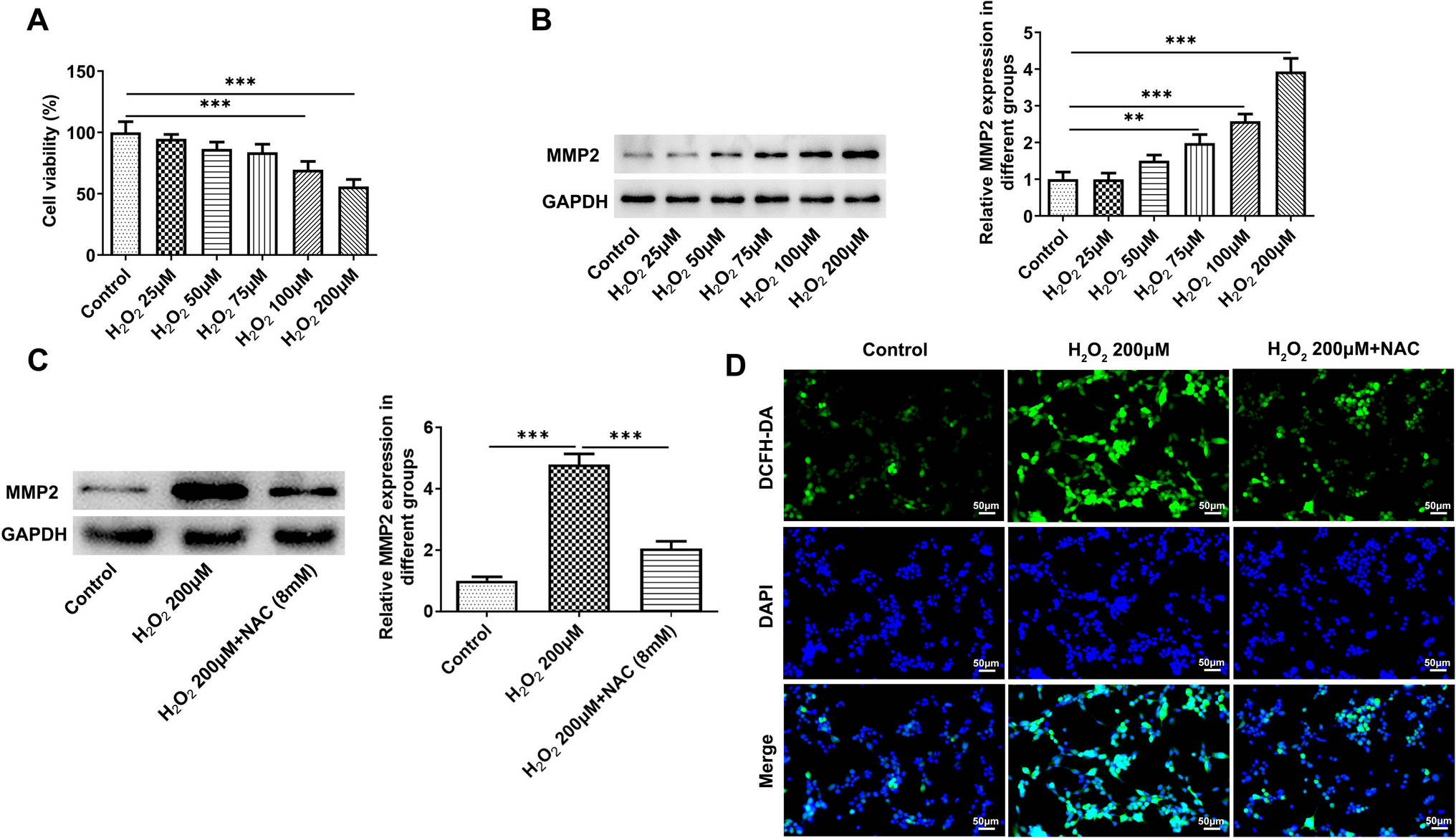
Cultivation of MLE-12 cells was carried out in 96-well plates for 48 h at 5 × 103 cells per well. After that, 10 µl of MTT (5 mg/mL) was added to each well and the mixture was incubated for 2 h. Afterwards, DMSO (100 µl per well) was used to replace the MTT reagent for dissolving the crystals. The mixture was shaken for 10 min at room temperature, followed by measurement of absorbance at 490 nm using a microplate reader (Bio-Tek, Vermont, USA).
Detection of malondialdehyde (MDA), glutathione (GSH), and Fe2+ levels
Determination of Fe2+ levels in mouse lung tissues and MLE-12 cells was done as per the instructions of the iron assay kit (Sigma). Lipid peroxidation levels and GSH levels were assayed according to the instructions of the MDA Assay Kit (Beyotime) and the GSH Assay Kit (Beyotime).
Ferroptosis was observed by transmission electron microscopy
The morphology of the cells was directly observed by transmission electron microscopy. The mitochondria became smaller and the mitochondrial membrane density was larger when ferroptosis occurred.
Cell count and protein concentration detection in BALF
To prepare BALF, the lungs were irrigated with sterile saline 3 times through endotracheal intubation. After centrifugation, the supernatant was stored at -20℃. The precipitated cells were resuspended in saline and the total cell count was measured using a hemocytometer. The percentage of neutrophils was measured by Wright-Giemsa staining. The protein concentration of BALF was determined with BCA protein assay kit (Biyuntian, Jiangsu).
Western blot
RIPA buffer containing protease inhibitors was used to extract total proteins from homogenized mouse lung tissues and MLE-12. Forty to eighty micrograms of proteins were separated on 10% SDS-PAGE gels and then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Burlington, USA). Following blocking, PVDF membranes were incubated for 12 h at 4 °C with the indicated primary antibodies (Abcam, Cambridge, UK): GPX4 (1:1000, #ab125066), SLC3A2 (1:1000, # ab303510), LC-3II/I (0.5 µg/mL, #ab62721), p62 (1:1000, # ab109012), FTH1 (1:1000, #ab183781), NCOA4 (1:1000, #ab314553), YTHDC1 (1:1000, # ab259990), Angptl4 (1:1000, #ab196746) and GAPDH (1:10000, #ab181602). An incubation of the PVDF membrane with goat anti-rabbit secondary antibody (1:2000, #ab6721) was then performed for 50 min. Density of protein bands was quantified using the Alphalmager™ 2000 Imaging System (Alpha Innotech, San Leandro, USA).
Distribution of ferrous iron in lysosomes
After incubation of MLE-12 cells in 96-well plates, the medium was removed, followed by the addition of 50 nM LysoTracker Green (Beyotime) and 1 mM FerroOrange (Dojindo, Japan). Co-staining was performed for 30 min, followed by replacing the previous solution with fresh PBS (Beyotime). After rinsing with PBS, samples were incubated with secondary antibodies rabbit anti-mouse IgG (Invitrogen) or goat anti-rabbit IgG (Invitrogen). Cell nuclei were stained with DAPI (Beyotime). Cell photographs were taken with an inverted microscope (Olympus, Tokyo, Japan). Quantification was performed by the software Image-Pro Plus 6.0.
Colocalization of LC3 and ferritin
Transfection of MLE-12 cells with LC3-GFP (Beyotime) for 24 h, followed by stimulation of cells with LPS for 24 h. Cells were permeabilized with 0.03% Triton X-100 (Beyotime) for 60 min, then fixed and blocked with 0.1% BSA (Beyotime) for 1 h. The primary anti-ferritin (Abcam, 1:100, ab75973) and secondary antibodies were applied in turn. MLE-12 cells were evaluated by fluorescence microscopy (Nikon TE-2000, Tokyo, Japan).
For immunofluorescence (IF) staining in lung tissues, the samples were fixed in 4% paraformaldehyde (Beyotime) for 1 h and permeabilized with 0.1% Triton X-100 (Beyotime) for 1 h. After incubation with primary antibodies including anti-LC3 (Affinity Biosciences, 1:100, AF5402) and anti-ferritin (Abcam, 1:100, ab75973), the samples were incubated with secondary antibodies. The samples were imaged with an inverted microscope (Olympus).
Animal models and grouping
Adenoviruses harboring sh-Mir22hg (Ad-sh-Mir22hg) was obtained from FitGene Biotechnology Co. (Guangzhou, China). Ad containing NC was utilized as the negative control (Ad-NC). All animal experiments were approved by the Institutional Animal Care and Use Committee of SSL Central Hospital of Dongguan City and were performed strictly according to national guidelines. Sepsis mouse models were established as previously described (Xu et al. 2020). After one week of acclimatization feeding, male C57BL/6J mice (6–8 weeks old and 18–21 g weight) were randomly divided into 3 groups, including ctrl + ad-NC, LPS + ad-NC and LPS + ad-sh-Mir22hg. Briefly, mice were subjected to sodium pentobarbital (50 mg/Kg body weight) anesthesia, followed by intraperitoneal injection of 15 mg/Kg LPS (Sigma). An equal amount of PBS was injected intraperitoneally into control mice. Other mice were injected with ad-sh-Mir22hg or ad-NC by the tail vein. All mice were sacrificed and lung tissues were isolated for subsequent analysis.
Hematoxylin-eosin (HE) staining
Isolated lung samples were fixed with 4% paraformaldehyde for 48 h, followed by dehydration with different concentrations (70-100%) of ethanol solution for 40 min. After paraffin embedding, the samples were cut into 4-mm slices, which were then deparaffinized in xylene, anhydrous ethanol and alcohol. Slides were stained with hematoxylin for 5 min, followed by eosin staining for 1–3 min.
Quantitative real-time PCR (qPCR)
RNA samples from cells were extracted with TRIzol® reagent (Life technologies, Carlsbad, CA, USA) according to the method provided by the manufacturer. First-strand cDNA was synthesized using Hiscript III reverse transcriptase (Vazyme, Nanjing, China). Relative RNA levels were determined by qPCR on a 7900 Real-Time PCR (Applied Biosystems, USA) system along with ChamQ SYBR qPCR Master Mix (Vazyme). GAPDH was used as an internal control to quantify mRNA levels of genes. Relative levels of mRNA were calculated using the comparative CT (2−ΔΔCT) method.
Subcellular fractionation
Cytoplasmic and nuclear RNA fractions from MLE-12 cells were prepared and collected according to the instructions of the PARIS™ RNA Nuclear/Cytoplasmic Isolation Kit (Life technologies). The levels of Mir22hg were determined by qPCR. U6 was used as a nuclear endogenous control. GAPDH was used as a cytoplasmic control.
RNA-FISH assays
The cells were fixed with 4% paraformaldehyde and then permeabilized with 0.1% Triton X-100. After washing, the cells were incubated with the probe overnight at 37◦C. DAPI was used for nuclear staining. The FISH probe was prelabeled at the 5 -end with Alexa Fluor 546 NHS ester. Fluorescence was detected using a DMI8 microscope (Leica).
In vitro pull-down assay with biotinylated RNA
The m6A sites in Angptl4 was mutated (mut). Full-length Mir22hg (F1), truncated Mir22hg (F2-F9), full-length Angptl4 wild type (WT) and its m6A mut sequence were biotinylated in vitro using the AmpliScribe T7-flash Biotin-RNA Transcription Kit according to the manufacturer’s instructions. Protein samples of MLE-12 cells were incubated with streptomycin beads (Invitrogen) preincubated with a biotinylated probe for 1 h. The complexes were precipitated, washed four times, and analyzed by western blot.
RNA-binding protein immunoprecipitation (RIP)
Using the Magna RIP Kit (Millipore), we performed RIP assays by following the manufacture’s instructions. Cells lysed with RIP lysis buffer, and the extracts were mixed with magnetic beads and incubated with primary antibodies against IgG (ab172730, 1:100, Abcam) or YTHDC1 (ab259990, 1:50, Abcam) for 2 h at 4 °C. RNA was then purified with TRIzol (Life technologies) and quantified by qPCR analysis.
Methylated RNA immunoprecipitation (MeRIP)
After 48 h transfection, total RNA (300ug/time) was extracted and MeRIP experiment was performed using the Magna MeRIP Kit (Millipore, Burlington, MA, USA) according to the manufacture’s instructions. In brief, the cells were lysed in a MeRIP lysis buffer, and magnetic beads were conjugated with a mouse antibody against m6A (ab208577, 1:50, Abcam) or IgG (ab172730, 1:100, Abcam) for 2 h at 4 °C. Subsequently, the obtained RNA was assessed by qPCR.
RNA stability assay
Following overnight incubation of cells in 6-well plates, 5 µg/mL of actinomycin D (Sigma) was added for inhibiting gene transcription. After that, RNA was then extracted and subjected to qPCR analysis.
Statistical analysis
Results are reported as mean ± standard deviation (SD). All experiments were repeated at least three times, each time with three technical replicates. Significant differences between two groups that conformed to normally distributed data were tested using Student’s t-test. For comparisons of more than two groups, ANOVA with Tukey’s post hoc test was used to calculate p-values. P < 0.05 was considered statistically significant, and p < 0.01 and p < 0.001 were considered statistically highly significant.

Comments (0)