
Remember me







This prospective, single-center, parallel-group trial is being conducted at the Peking University Third Hospital. This trial is registered with ClinicalTrials.gov (NCT05914857). The study methods have been reported in accordance with the CONsolidated Standards Of Reporting Trials (CONSORT) statement (Supplementary Material) [33, 34]. Figure 1 shows the flow diagram of the study. This study protocol is reported according to the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines for defining items of the clinical trial (Supplementary Material) [35].
Fig. 1
Enrollment and follow-up: CONSORT flow diagram describing anticipated progress of participants through the trial
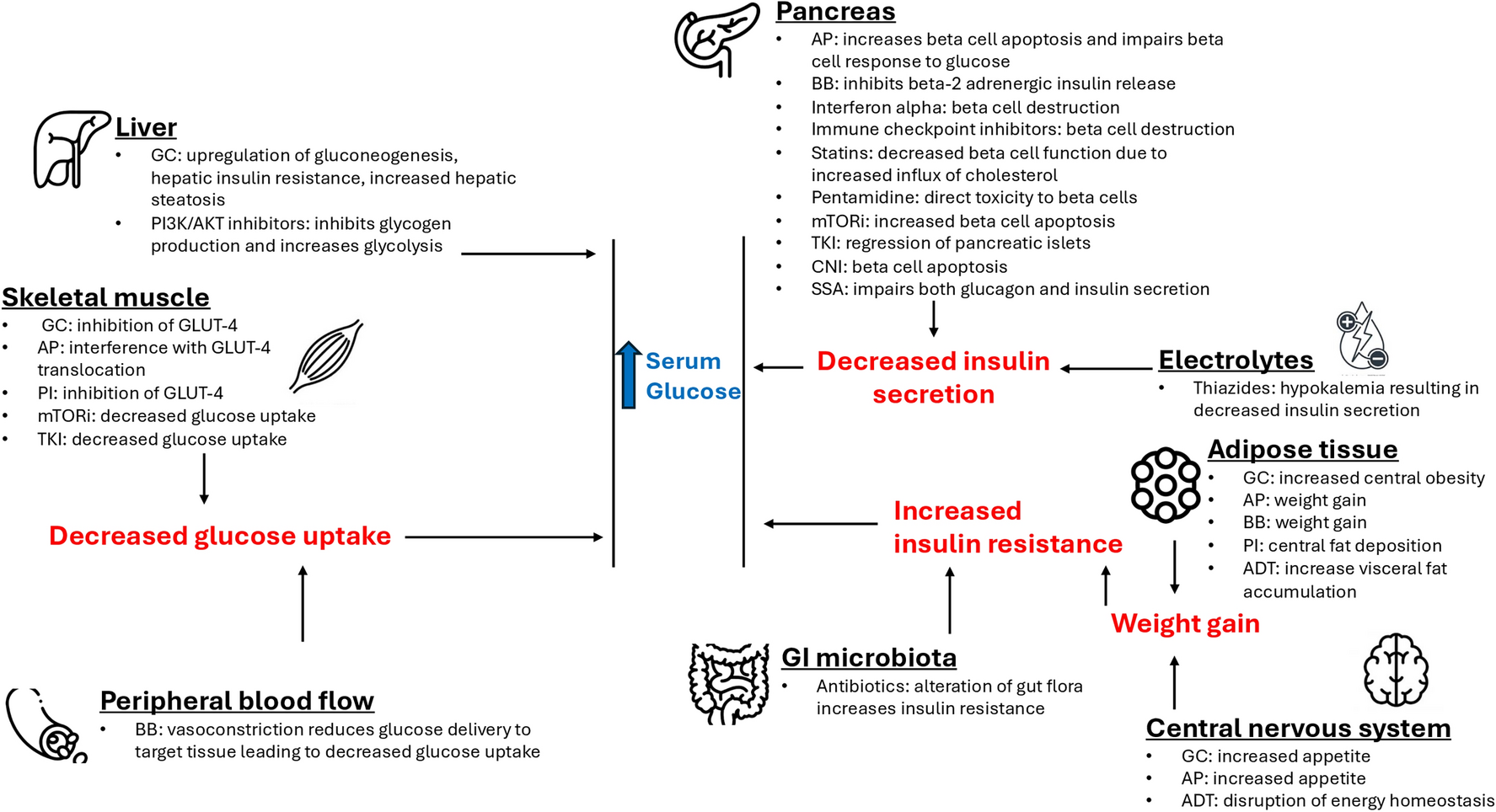
Trial Design and Ethical ApprovalsA total of 240 people with prediabetes who meet the predefined inclusion and exclusion criteria will be randomly assigned to the intervention or control group in a 1:1 ratio. The intervention group will receive dapagliflozin (10 mg/day) along with lifestyle education, while the control group will undergo lifestyle education alone (with male/female = 1:1 in each group). The trial will span a duration of 12 weeks to closely monitor changes in metabolic parameters, including glucose levels. The study is being conducted in accordance with the Declaration of Helsinki, and ethical approval has been obtained from the Peking University Third Hospital Medical Science Research Ethics Committee (reference number CN-032), with the approval number IRB00006761-M2022520. Any study discontinuation, completion, or changes in conduct (e.g., protocol revisions) must be promptly reported to the committee for approval. Immediate notification is required if changes are made to mitigate risks to subjects.
Participant EligibilityEligible participants will meet the following criteria:
1.Age between 18 and 65 years
2.Diagnosed with prediabetes based on the combined criteria of American Diabetes Association and World Health Organization within the last 6 months:
Fasting blood glucose between 6.1 and 7.0 mmol/L (110–125 mg/dL)
2-h plasma glucose (2 h-PG) on a 75-g oral glucose tolerance test (OGTT) between 7.8 and 11.1 mmol/L (140–199 mg/dL)
HbA1c between 5.7% and 6.4% (39–46 mmol/mol)
Fulfillment of at least two criteria above is required for diagnosis. If only one criterion is met initially, the diagnosis must be confirmed by repeating the test on a different day.
Fasting blood glucose and HbA1c levels will be assessed following an overnight 8–12 h fasting, usually conducted in the morning. 2 h-PG on a 75-g OGTT will be measured 2 h after ingesting 75 g anhydrous glucose powder dissolved in 300 mL water, following overnight fasting.
Participants will be recruited by the researchers, and study documentation will be provided in advance. Those interested in participating will be scheduled for an appointment to discuss the study and provided written informed consent before engaging in any trial-related activities.
Participant Exclusion and Discontinuation CriteriaExclusion Criteria 1.Diagnosed with diabetes
2.Taking antidiabetic, lipid-affecting, or weight-loss medications within the 6 months prior to screening (including traditional Chinese medicine, and excluding thiazide diuretics at a daily dose of ≤ 12.5 mg)
3.Patients with acute infection, surgery, acute alcoholism, or mental illness
4.Patients with liver and kidney dysfunction, severe chronic gastrointestinal disease, uncontrolled thyroid disease, cancer, or on ventilator support
5.Systolic blood pressure ≥ 180 mmHg (1 mmHg = 0.133 kPa) or diastolic blood pressure ≥ 110 mmHg at screening
6.Electrocardiogram within 12 weeks before screening indicating arrhythmia requiring urgent diagnosis or treatment (e.g., clinically newly identified severe arrhythmia or conduction disturbance), myocardial infarction, unstable angina, or stroke requiring cardiovascular and cerebrovascular intervention
7.A history of traumatic amputation within the past year, or active skin ulcers, osteomyelitis, gangrene, or critical lower limb ischemia within the past 6 months
8.Enrolled in drug/device clinical studies (including vaccines) within 12 weeks before screening
9.Pregnant or lactating individuals, or those planning a pregnancy
10.Allergic constitution or multidrug allergy
11.Receiving bariatric surgery within the past 2 years
12.Receiving unsatisfactory pre-trial adherence evaluations that might hinder participants from following and completing the trial
Discontinuation Criteria 1.Pregnancy
2.Inability to follow lifestyle instructions as per protocol
3.Patients with severe cardiovascular events or pulmonary embolism
4.Loss of contact with participants
5.Subjects request early withdrawal from the study (for reasons other than adverse reactions or a lack of efficacy)
6.Other reasons (recorded in the case report form)
A candidate participant who is already engaging in exercise or following a specific diet may still be eligible for participation in the study. Their exercise and dietary patterns will be systematically monitored and documented before and throughout the study. These data will be analyzed in the results after the completion of all recruitments.
Patient WithdrawalPatients may opt to discontinue their participation in the trial at any point. Withdrawal from the trial is permitted and may be advised under certain circumstances, including adverse events, safety concerns, or a lack of adherence to the trial protocol.
Eliminated CasesParticipants who do not complete the trial as planned and have incomplete observation records are eliminated. Subjects who discontinue the trial as a result of serious adverse reactions should be classified as adverse reaction cases and should not be included in the per-protocol (PP) analysis.
Design of the InterventionParticipants are stratified by gender and then randomly allocated to the intervention and control groups. The intervention group receives dapagliflozin 10 mg/day alongside lifestyle education, while the control group is given lifestyle education alone. Participants will be assigned randomly to either the intervention or control group by employing a random number table.
Dapagliflozin (Forxiga®) will be administered orally as 10-mg tablets, once daily.
Lifestyle EducationRecommended lifestyle interventions include a sensible diet, controlling calorie intake (reducing total dietary calories by at least 400–500 kcal per day), and engaging in moderate-to-high intensity physical activity for more than 30 min per day, according to the Intervention for Adults with Pre-diabetes: A Chinese Expert Consensus [14]. We will provide lifestyle education, including instructions on physical activity and diet intake, at the start of the intervention. We will follow up their adherence to lifestyle modifications during two phone follow-ups and the end visit. Study participants should be instructed not to donate blood or blood products during the study.
Physical activity: For optimal physical fitness, it is highly recommended to incorporate a combination of aerobic and resistance exercises daily for a minimum of 30 min. Aerobic exercises, including brisk walking, jogging, cycling, and swimming, should be performed at least three times weekly. Resistance exercises, which entail the utilization of resistance training equipment or free weights (such as dumbbells and barbells), should be integrated into the exercise regimen at least twice a week, in conjunction with regular aerobic exercise.
Dietary intervention: The daily total energy requirement is recommended to comprise 45–60% from carbohydrates, 25–35% from fats, and 15–20% from protein. The daily dietary energy intake is advised to be reduced by a minimum of 300 kcal. Furthermore, it is crucial to limit the intake of saturated fatty acids to less than 30% of the total fat intake and restrict daily salt intake to no more than 6 g. It is not advisable to consume alcohol; if consumed, it must be included in the total energy intake (7 kcal/g).
Adherence EvaluationPre-trial AssessmentDuring the initial visit, subjects will be evaluated for various factors to select participants with good adherence. These factors include their occupation, regularity of daily life (potential hindrance due to occupational commitments impacting exercise and dietary goals), awareness of prediabetic state and its risks, inclination towards improving their current health condition, and ease of communication. Individuals with factors suggesting they might struggle with adherence will be excluded.
In-trial AssessmentDuring the period of the trial, patients will receive telephone follow-ups to assess their adherence to study interventions. Key self-reported measures include medication adherence, frequency, and reasons for non-adherence; weekly physical exercise frequency, duration, and type; and dietary habits, encompassing frequency of dining out, consumption of high-fat/oil foods, and overall diet maintenance. Patients will self-report over the telephone how many pills they have left, which will be cross-referenced with expected amounts to assess medication adherence.
For some eligible patients, objective measures will be integrated: data from a designated physical activity app to assess exercise habits, and meal photographs to evaluate dietary adherence.
These multifaceted evaluations ensure a robust assessment of adherence, reinforcing the reliability and validity of study results.
The gathered adherence data will contribute to a comprehensive understanding of participants’ adherence to the study interventions. This methodology ensures a rigorous evaluation of adherence throughout the trial, contributing to the reliability and validity of the study findings.
Baseline Measurements and RandomizationBaseline assessments encompass both clinical and laboratory measurements. We will record age, sex, BMI, waist circumference, and blood pressure. At the start and the end of the trial, we will collect serum and urine samples to evaluate glucose and other metabolism parameters and collect semen samples for evaluation safety of male reproductive function.
The eligible participants will be allocated concealed and randomized in a 1:1 ratio. Randomization will be stratified by gender. Prior to this, a predetermined list comprising 120 research subject numbers was established separately for each gender. A random number table method was employed to generate random digits. Subjects were allocated into one of two groups, intervention group and control group, on the basis of the parity of the generated random digits—subjects assigned odd numbers were placed in the intervention group, while those with even numbers were assigned to the control group.
The assignment of medications was predetermined for the groups: the intervention group received dapagliflozin 10 mg/day alongside lifestyle education, while the control group is given lifestyle education. Subsequently, research personnel who were not part of the trial recruiters or intervention allocators preserved a record of the random allocation scheme within sealed opaque envelopes.
Clinical MeasuresMeasurements will be performed by trained study physicians, following standard protocols. Clinical measures will encompass BMI, waist circumference, blood pressure, along with participants’ medical history and medication intake. Participants will be measured barefoot and in light clothing. Weight will be recorded to the nearest 100 g using a calibrated electronic scale. Height will be assessed to the nearest 0.1 cm with a calibrated wall-mounted stadiometer. BMI (kg/m2) will be calculated as weight divided by square height. Blood pressure (mmHg) will be taken once seated using a calibrated automatic oscillometric sphygmomanometer and systolic and diastolic blood pressure will be recorded. In the standing position with feet 25–30 cm apart, waist circumference is measured at the midpoint between the lowest rib margin and the anterior superior iliac spine and will be measured to the nearest 0.1 cm using a non-stretchable fiberglass tape. Comprehensive patient data collection will comprise demographic and medical history, inclusive of age, gender, smoking history, history of cardiovascular disease, biochemical hypoglycemia, severe liver disease, malignancy, stroke, genitourinary infection, and usage of hypoglycemic and/or weight-loss drugs.
Laboratory MeasuresLaboratory measurements will be conducted at the study initiation (before medication intake) and the study end (following a 12-week intervention period). The measurements included blood glucose and other metabolic parameters.
Venous blood samples will be collected from all participants. Whole blood samples will be used to ascertain the complete blood count (automated hematology analyzer) and to assess HbA1c (ion-exchange high-performance liquid chromatography method). Plasma will be used to evaluate blood glucose level, including fasting blood glucose, OGTT measurements at 0.5 h, 1 h, and 2 h glucose (glucose oxidase method). Serum will be collected for other biochemistry tests. Fasting insulin levels alongside insulin at each OGTT time point will be measured (the Siemens Insulin Assay Kit, chemiluminescence assay). The lipid profile includes total cholesterol, high-density lipoprotein cholesterol, low-density lipoprotein cholesterol, and triglyceride (turbidimetric immunoassay). Liver function includes alanine aminotransferase (lactate dehydrogenase method) and aspartate aminotransferase (malate dehydrogenase method). Kidney function will be reflected by blood creatinine levels (picric acid method). Sex hormones including follicle-stimulating hormone, luteinizing hormone, prolactin, estradiol, progesterone, and testosterone will be quantified (Roche Diagnostics; chemiluminescent assay). The blood samples for sex hormone detection in women should be collected under fasting conditions between 8:00 and 10:00 AM on days 2 to 5 of the menstrual cycle. For those experiencing amenorrhea exceeding 60 days or postmenopausal individuals, blood samples should be obtained before 10:00 AM on any day.
In terms of urinary evaluations, participants’ urine samples will be subjected to routine testing (automated urinalysis instrument). Urine creatinine levels will be gauged (picric acid method), and albumin/creatinine ratio levels will be measured to monitor for early signs of proteinuria.
Fecal samples from participants will undergo routine examinations under microscopic observations.
For male volunteers aged between 18 and 50 years, semen samples will be analyzed with the sperm quality analyzer and computer-aided semen analysis system. After collection, these samples will undergo incubation at 37 °C for 0.5–1 h and will be checked for the absence of liquefaction before centralized assessment at the Department of Reproduction Center at Peking University Third Hospital.
Upon collection, all specimens—with the exception of semen samples—will be processed promptly within 2 h at the centralized laboratory of the Department of Laboratory Medicine at Peking University Third Hospital. Following their analyses, all samples, including the semen samples, will be stored in the Peking University Third Hospital Biobank for a maximum duration of 15 years, subject to ethical board clearance. At the conclusion of this period, any remaining samples will be disposed in accordance with relevant guidelines.
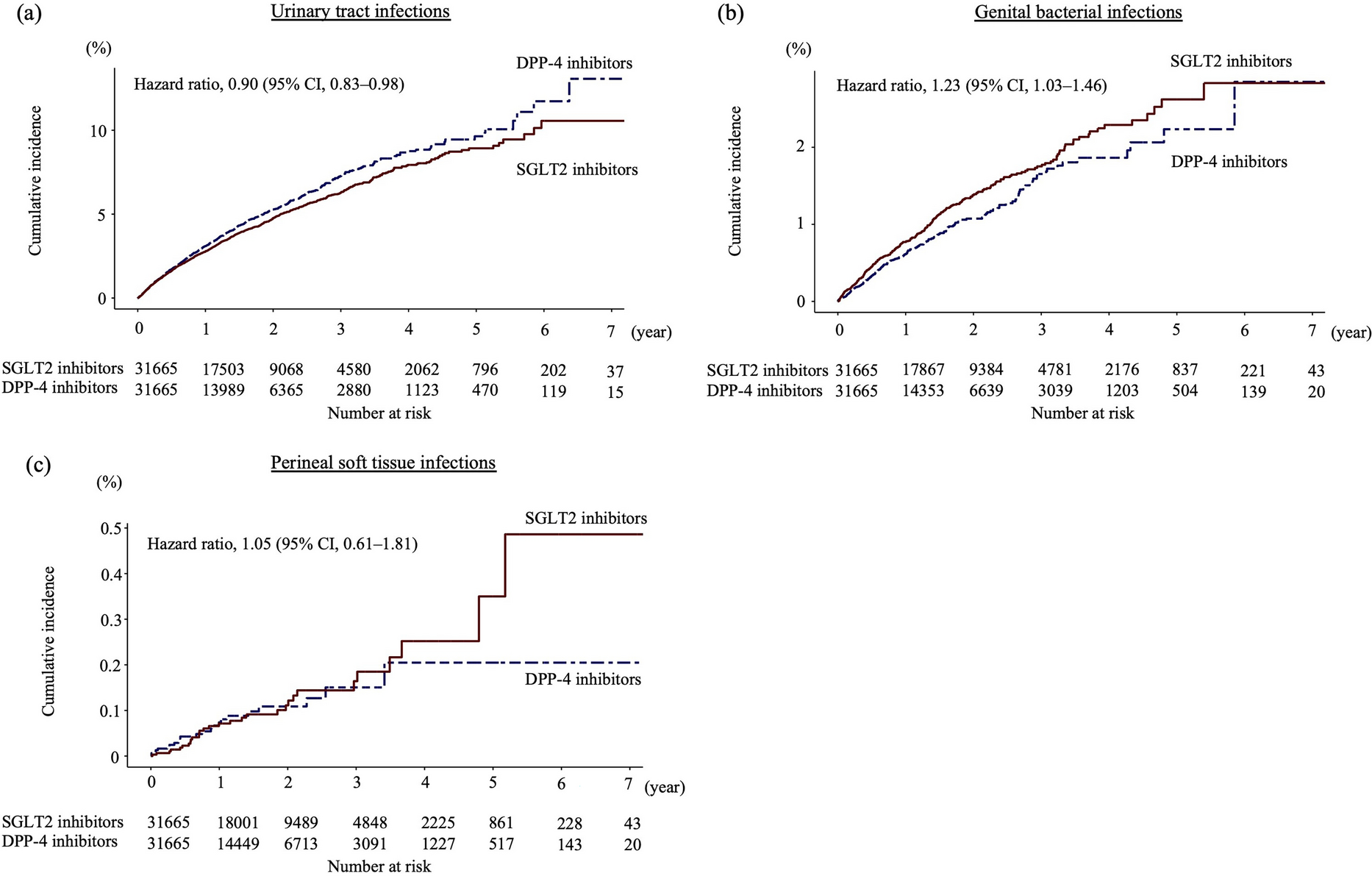
Intervention VisitsRegular telephone follow-ups are carried out at week 4 and week 8 to assess medication adherence, potential adverse drug reactions, and physical activity. Table 1 delineates the specific content arrangements within the follow-up procedures. Figure 2 provides a comprehensive visualization of the study’s sequential assessments and informational components.
Table 1 Patient evaluation follow-up scheduleFig. 2
Protocol summary for the randomized controlled trial of dapagliflozin in subjects with prediabetes
Study OutcomesPrimary OutcomeThe primary efficacy endpoint is the change of 2 h-PG during OGTT from baseline to study end (12 weeks).
There are several reasons. First, we are monitoring fasting blood glucose, 2 h-PG, and HbA1c levels to assess the potential of SGLT2 inhibitors in restoring normal glycemic control among individuals with prediabetes. It is worth noting that individuals with IGT outnumber those with IFG in the prediabetic population [36]. Additionally, previous studies have shown that 2 h-PG serves as a strong predictor of cardiovascular events in individuals with coronary heart disease, underscoring its clinical significance [37]. Moreover, the 2 h-PG is widely used as a surrogate marker for assessing glycemic control and is recommended in guidelines for evaluating prediabetes and diabetes [14, 38, 39]. Therefore, we have chosen 2 h-PG level during OGTT as the primary endpoint.
Secondary OutcomesSecondary efficacy outcomes include changes in fasting blood glucose, HbA1c, BMI, waist circumference, fasting insulin, OGTT-2 h insulin, blood lipid profile (total cholesterol, high-density lipoprotein cholesterol, low-density lipoprotein cholesterol, and triglyceride), and blood pressure (including diastolic blood pressure and systolic blood pressure).
The safety profile is evaluated through changes in liver function (alanine aminotransferase and aspartate aminotransferase levels), kidney function (creatinine, calculated estimated glomerular filtration rate, and albumin/creatinine ratio), urine routine tests, sex hormones, sperm-related parameters (count and function), and self-reported adverse events.
Quality ControlTo maintain the highest quality in data collection and analysis throughout the study, we will implement comprehensive quality control strategies. These strategies will focus on reinforcing standardized procedures and minimizing both inter- and intraobserver variations.
(a)Ethical approval: We submit the research plan to the Medical Science Research Ethics Committee of Peking University Third Hospital for thorough review and approval to ensure the study complies with all ethical standards and regulations.
(b)Researcher training: Prior to data collection, all researchers will undergo comprehensive training to ensure accurate and consistent recording and evaluation of each case. In preparation for this, we design a case report form informed by extensive consultations with experts and a thorough review of relevant literature, intending to ensure data accuracy and reliability.
(c)Participant retention strategies: We recognize the importance of minimizing participant attrition to maintain the robustness of our results. Therefore, we will implement public awareness and education campaigns targeting our participants to increase their understanding of the study’s importance and reduce the rate of dropout.
(d)Data management: To ensure the accuracy and reliability of all recorded outcomes, this study will implement a double data entry method, aiming to elevate the standards of data quality.
Sample Size EstimationSample size estimation for this trial was conducted on the basis of the primary efficacy endpoint: the change of 2 h-PG during OGTT from baseline to 12-week intervention. Both treatment and control groups are expected to experience a reduction in 2 h-PG levels. However, the dapagliflozin group is anticipated to show a greater reduction by 0.86 mmol/L compared to the control group. This estimate is adapted from previous RCTs involving metformin as a reference therapeutic agent [40]. Gender stratification will maintain a 1:1 male-to-female ratio, facilitating the exploration of potential gender-based differences in adverse effects, particularly those related to the genitourinary system. Six sex hormone tests and semen analysis are included in the study’s metrics to monitor these gender-specific adverse effects, as supported by relevant literature [41].
For sample size calculation, we employ the following formula for a two-sample t test:
where Zα/2 = 1.96 for a two-sided α level of 0.05, Zβ = 0.84 for a power of 80%, Δ = 0.86 mmol/L (expected difference), and σ = 1.52 (standard deviation). The formula yields n = 49 participants per group.
Considering a dropout rate of 15%, the sample size per group needs adjustment to accommodate potential participant attrition. The adjustment increases the necessary number of participants per group from 49 to 58. To ensure robustness, we will include 60 participants for each gender in each group. Consequently, the adjusted total sample size for the trial will be 240 participants, comprising 60 men and 60 women in both the intervention and control groups.
Recruitment advertisements targeting potential participants will be disseminated through various channels, ensuring the attainment of the predetermined sample size necessary to maintain the study’s validity and robustness.
Statistical AnalysisStatistical analysis will be performed using SPSS 25.0 software. The independent sample t test, chi-square test, Spearman’s correlation analysis, and binary logistic regression analysis will be used for comparative and correlation analysis respectively. A P value of less than or equal to 0.05 will be considered statistically significant.
We will conduct an intention-to-treat (ITT) analysis, where all participants will be included in the analysis according to their randomization group, regardless of whether they received the intervention or completed the study. We will also conduct a PP analysis, which will include only participants who completed the study as per the protocol without major deviations.
For handling missing data, we will employ multiple imputation techniques, which allow for the estimation of missing values by leveraging observed data and making informed assumptions regarding the reasons for data absence. This approach is particularly advantageous in scenarios where covariates themselves have missing values or when covariates that change over time can predict future instances of missing data [42]. Imputations will be performed with the Multiple Imputation by Chained Equations (MICE) package in R software [43].
Trial StatusThe trial is currently in the recruitment phase. We expect to complete patient enrollment intervention and follow-up period by March 2025, adhering to the original schedule.
Comments (0)