
Remember me

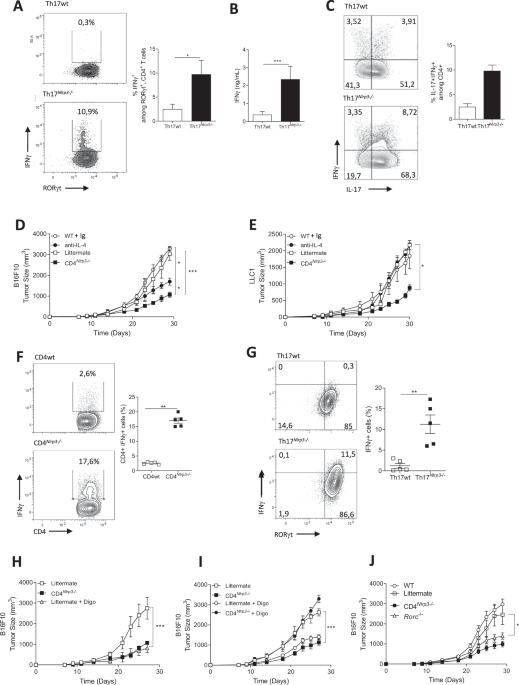




Phgdhfl/fl mice were crossed with Cx3cr1cre/+ mice to obtain Phgdhfl/flCx3cr1cre/+ (denoted as Phgdhfl/flCx3cr1-Cre) or Phgdhfl/flCx3cr1+/+ (denoted as Phgdhfl/fl) littermates. All mice in the study were bred on a C57BL/6 background and housed in conventional cages with access to a standard diet and water ad libitum at a constant ambient temperature and a 12-hour light cycle. For the tumor experiments, first, AE17 cells were injected intraperitoneally (i.p.) in mice to promote tumor growth. These recultured and STR-checked AE17 cells were subcutaneously injected (1 ×106 in 100 μL of serum-free RPMI plus 25% Matrigel) into the right flank of 8- to 12-week-old Phgdhfl/fl and Phgdhfl/flCx3cr1-Cre mice. Tumor size was monitored at regular intervals of 2–3 days by measuring the length and width with a caliper. The tumor volume was calculated using the following formula: tumor volume = (length × width2)/2. When the tumors reached a size of 500–800 mm3, the animals were sacrificed by cervical dislocation, and the tumors were collected for further analysis. The experiments were repeated three times, with at least three mice per condition injected with tumor cells in each experiment. The therapeutic experiments conducted in this study were approved by the official Austrian ethics committee for animal experiments (ethics approval number: GZ 66.009/0134-V/3b/2019). Mice in individual experiments were sex- and age-matched.
Cell cultureThe human lung cancer cell Line A549, the human breast cancer cell line MDA-MB-231, the human colon cancer cell line HCT116, mouse RAW 264.7 macrophages, and the mouse fibroblast Line L929 were originally obtained from the ATCC. The mouse mesothelioma cell line AE17 was originally obtained from Sigma. All cancer cell lines and L929 cells were grown in Dulbecco′s modified Eagle′s medium (DMEM) high glucose mixture supplemented with 10% heat-inactivated bovine serum (FBS), 2 mM L-glutamine, 100 U/mL penicillin and 0.1 mg/mL streptomycin (complete DMEM). For metabolomic analysis, dialyzed heat-inactivated FBS was used. All the cell lines used in the study were confirmed to be free of mycoplasma contamination.
Conditioned media preparationA total of 0.25 ×106 cancer cells were seeded in 15 mL of complete DMEM. After 24 h, the tumor-conditioned medium (TCM) was collected and centrifuged at 400 × g for 4 min. The supernatant was filtered through a 0.22-µm filter to eliminate debris and stored at −80 °C before use. For the L929-conditioned medium, a total of 17 × 106 L929 cells were cultured in 40 mL of complete DMEM in 175 cm2 flasks. After 7 days, the L929 conditioned medium was collected, filtered (0.22 µm), aliquoted and stored at −20 °C until use.
Macrophage isolation and in vitro polarizationBMDMs were generated as previously described [53]. Briefly, bone marrow cells were obtained by flushing the femurs and tibias of wild-type (WT), Phgdhfl/fl or Phgdhfl/flCx3cr1-Cre mice aged 6 to 8 weeks. After erythrocyte lysis with ammonium chloride-potassium (ACK) lysis buffer (0.15 M NH4Cl, 10 mM KHCO3 and 0.1 mM EDTA) for 5 min at room temperature (RT), the cells were washed twice with ice-cold phosphate-buffered saline (PBS), and the bone marrow cell suspension was cultured in BMDM differentiation medium (high glucose DMEM, 10% heat-inactivated FBS, 2 mM L-glutamine, 100 U/ml penicillin and 0.1 mg/ml streptomycin supplemented with 20% L929-conditioned supernatant) for 7 days. Fresh medium was added every 2–3 days. On Day 7, differentiated BMDMs (96% of the cells were positive for F4/80 and CD11b) were harvested and seeded in complete DMEM for different experiments.
To induce in vitro polarization, BMDMs were plated at a density of 1 ×106 cells/well in six-well plates with 2 mL of DMEM and allowed to adhere overnight. The media was then replaced with TCM supplemented with 25 ng/mL IL-4 or 100 ng/mL LPS. After 4, 8 or 24 h of stimulation, the macrophages were collected for RNA or protein extraction. For tumor coculture experiments, AE17 cells were plated at a density of 3 × 105 cells in a 24-well plate and allowed to attach overnight. Then, 4 × 105 macrophages were seeded onto the cell inserts (0.3 μm pore size) for 10 min before they were added to the tumor cells. After stimulation with 100 ng/mL LPS for 24 h, the macrophages were collected for RNA extraction. For several experiments, macrophages were cultured with IL-4 in the presence or absence of 25 μM WQ-2101 (SML1970; Sigma), 0.4 mM serine (Sigma), 1 mM formate (Sigma) or 1 mM DM-αKG (Santa Cruz; sc-211344) for 24 h, after which the cells were harvested for further experiments as indicated.
Flow cytometry of cells from tumor tissueTumors were excised, minced and dissociated in digestion buffer (1 mg/mL collagenase VIII and 1 mg/mL DNase I in PBS). The tissue was incubated for 30 min at 37 °C with agitation and then filtered through a 70 μm Falcon cell strainer to remove undigested tumor tissues. Red blood cells were lysed with ACK lysis buffer. Viable cells were counted after resuspension in WB buffer (5% FCS, 5 mM EDTA, and 20 μg/mL DNase I in PBS without Ca2+/Mg2+). Subsequently, 2–4 ×106 cells in 100 μL of WB buffer were blocked with Fc blocking solution (anti-mouse CD16/CD32, 1:1000) for 10 min on ice. The cell suspension was then divided into two parts for macrophage staining and T-cell staining. For macrophage staining, the cell suspension was incubated in WB buffer for 30 min on ice with antibodies specific for CD45 (1:100, 30-F11; BioLegend), F4/80 (1:100, BM8; BioLegend), CD11b (1:100, M1/70; BioLegend), CD206 (1:100, C068C2; BioLegend), I-A/I-E (1:100, M5/114.15.2; BioLegend) and PD-L1 (1:100, 10 F.9G2; BioLegend). For T-cell staining, the cell suspension was incubated in WB buffer for 30 min on ice with antibodies specific for CD45 (1:100, 30-F11; BioLegend), CD3 (1:100, 17A2; BioLegend), CD25 (1:100, PC61; BioLegend), CD4 (1:100, GK1.5; BioLegend), and CD8a (1:100, QA17A07; BioLegend). For dead cell staining, Zombie Aqua (BioLegend) was added to the T cells before the incubation of antibodies, and 7-AAD (BioLegend) was added to the macrophages after the incubation of antibodies. For intracellular protein staining, Perm Fix solution was used for fixation and permeabilization, followed by FOXP3 (1:100, MF-14, BioLegend) and IFN-γ (1:100, XMG1.2, BioLegend) staining for 30 min at RT. To improve IFN-γ staining, exocytosis was inhibited by keeping the cells in the presence of brefeldin A (10 μg/mL, BioLegend) during the isolation and staining procedure. The data were obtained on a BD LSRFortessaTM X-20 cell analyzer (Becton Dickinson) and processed using FlowJo v10.
TAM isolation using FACSTumors were isolated and digested as described above. The cell suspension was blocked with Fc blocking solution for 20 min on ice. The cells were subsequently incubated with fluorophore-conjugated antibodies against CD45, CD11b and F4/80 on ice in the dark for 30 min. After the incubation, the cells were washed with WB buffer and centrifuged at 300 ×g for 5 min. The cell pellet was resuspended in 500 µL of WB buffer containing 7-AAD and sorted using a BD FACSMelody™ Cell Sorter. The CD45+CD11b+F4/80+ cells were sorted directly into TRI reagent (Thermo Fisher Scientific; 15596026). Then, RNA was isolated from the sorted cells following the manufacturer’s instructions.
Cell cycle analysisAfter treatment, the BMDMs were incubated with 20 μM BrdU (B5002, Sigma) in fresh culture medium at 37 °C in a humidified atmosphere (5% CO2). After 2 h, the cells were washed twice with PBS (without Ca2+/Mg2+) and harvested. Subsequently, 1 × 106 cells were resuspended in cold (–20 °C) ethanol and fixed overnight at 4 °C. Afterward, the cells were washed and incubated in 2 M HCL containing 0.5% Triton X-100 at RT. After 30 min, the cells were washed again and incubated with 0.1 M Na2B4O7 for 10 min at RT. After another wash, the cell pellets were resuspended in anti-BrdU antibody solution (1:25, 3D4; BioLegend) and incubated for 30 min in the dark at RT. The cells were washed twice in cell staining buffer (BioLegend; 420201) and resuspended in 100 μL staining buffer containing 5 μL 7-AAD solution (BioLegend; 420403). After a 30 min incubation at RT in the dark, the cells were analyzed using a FACSAria II instrument (BD Biosciences).
Immunohistochemistry and immunofluorescence of tumor sectionsTumor tissues were fixed in 4% formaldehyde, dehydrated, embedded in paraffin, and sectioned into 4 μm thick slices. To quench endogenous peroxidases, the sections were incubated with H2O2 (0.3% v/v) in PBS for 10 min. Antigen retrieval was performed by boiling the sections in 10 mM citrate buffer (pH 6.0) for 30 min. Prior to antibody incubation, the sections were blocked with Ultra V Block solution (UltraVision LP, Thermo Fisher Scientific) for 1 hour at RT. The sections were then incubated with a Ki67 antibody (1:200, 28074-1-AP, Proteintech) in a humid chamber for 1 hour at RT. Antibody binding was detected using the UltraVision LP detection system according to the manufacturer’s instructions. Subsequently, the sections were developed with DAB reagent (Dako) and counterstained with hematoxylin for 30 s. In addition, H&E staining was performed as previously described [54]. The stained slides were scanned using a whole slide scanner (Pannoramic SCAN II, 3DHISTECH). Evaluation and quantification of Ki67-stained cells were performed using Definiens software.
For immunofluorescence (IF) staining, tumor tissues were embedded and frozen in OCT medium (Sakura Finetek) and sliced into 4 µm sections using a cryomicrotome (Cryostar NX70, Thermo Fisher Scientific). The cryo-slides were then defrosted and fixed with 4% PFA. To block nonspecific binding, the tissue slides were incubated with a blocking solution (5% goat serum, 1% bovine serum albumin and 0.1% or 0.3% Triton X-100 in PBS). Subsequently, the slides were incubated with antibodies against CD68 (1:800, D4B9C, CST) and p-S6 (1:300, 50. Ser 235/236, Santa Cruz) overnight or with PHGDH (1:400, D3D5E, CST) and CD3-FITC (1:400, 17A2, CST) for 1.5 h. The samples were washed three times with PBS and subsequently incubated with fluorescence-labeled secondary antibodies for 1 hour in the dark. After incubation, the slides were counterstained with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI; Sigma) for 10 min. The tissue slides were then mounted in nonhardening mounting medium (Vectashield Mounting Media, Vector). A confocal laser-scanning microscope (LSM 700, Carl Zeiss) equipped with a 63 Å/1.4 oil differential interference contrast M27 objective lens (Plan Apochromat, Carl Zeiss) was used to analyze the IF staining. The acquired images were processed using Zeiss ZEN 2.5 software.
RNA extraction and RT‒qPCRAfter treatment, BMDMs or TAMs were collected, washed twice with PBS and suspended directly in TRI reagent (Thermo Fisher Scientific). Total RNA was isolated following the manufacturer’s instructions. The concentration of RNA was determined using a Qubit 4 Fluorometer (Thermo Fisher Scientific), a Qubit RNA Broad Range Assay Kit (Invitrogen; Q10210), and a Nanodrop (Thermo Fisher Scientific). Reverse transcription of RNA was performed using the GoScriptTM Reverse Transcription Kit (Promega) with 1 μg of total RNA. The mRNA levels of the target genes were assessed using Luna Universal qPCR Master Mix (New England Biolab; M3003E) on a Bio-Rad CFX96 real-time system. The quantification of mRNA expression was performed via the comparative Ct (2-∆∆Ct) method. The Rps9 gene was used as the internal control. The primer pair sequences used are listed in Supplementary Table S2.
Enzyme-linked immunosorbent assay (ELISA)Differentiated BMDMs were seeded at a density of 1 ×106 cells per well in a 6-well plate overnight. The media was then replaced with AE17-TCM, A549-TCM, MDA-MB-231-TCM or HCT116-TCM. After 24 h, cell-free supernatants were collected. The levels of secreted TGF-β and VEGF in the supernatants were determined following the manufacturer’s instructions using a TGF-β ELISA kit (Invitrogen, BMS608-4) and a VEGF ELISA kit (Sigma, RAB0509-1KT). Respectively. The experiment was performed in three biological replicates.
Cell viability assayPhgdhfl/flCx3cr1-Cre and Phgdhfl/fl BMDMs were plated at a density of 1 ×104 cells/well into a 96-well plate for 24 h. The media were then replaced with 100 μL of fresh DMEM containing 10% CCK8 (MedChemExpress, HY-K0301) for 1 hour. The absorbance was measured at 450 nm by a microplate reader (Thermo Scientific).
Proliferation assay (CCK8)Phgdhfl/flCx3cr1-Cre, Phgdhfl/fl or WT BMDMs were seeded at a density of 5 ×103 cells/well into a 96-well plate and incubated overnight. The media were subsequently replaced with AE17-TCM, A549-TCM, or AE17-TCM supplemented with 100 nM rapamycin (MedChemExpress, HY-10219). CCK8 solution was added as described above, and the absorbance at 450 nm was measured every 24 h for a period of 3 days.
Western blot analysisMacrophages were homogenized in cold RIPA lysis buffer containing protease inhibitor cocktails (Sigma; 4693116001) and phosphatase PhosStop EASYPack cocktails (Roche; 4906837001) followed by sonication with a tip-probe sonicator on ice. The concentrations of the proteins were determined using the bicinchoninic acid (BCA) assay. Equal amounts of proteins were loaded onto 10% or 12% SDS‒PAGE gels, separated by electrophoresis, and then transferred onto polyvinylidene fluoride (PVDF) membranes. The membranes were subsequently incubated with primary antibodies. The protein bands were visualized using an enhanced chemiluminescence (ECL) system (Amersham Biosciences, Cytiva) and imaged using an iBright FL1500 Imaging System (Thermo Fisher). The primary antibodies used were anti-ARG1 (1:1000, sc-47715), p-ribosomal protein S6 (1:1000, sc-293144), anti-ribosomal protein S6 (1:1000, sc-74459), and anti-p-4E-BP1 (1:1000, sc-293124); anti-4E-BP1 (1:1000, sc-81149); anti-p-AKT1/2/3 (1:1000, sc-81433); and anti-AKT1/2/3 (1:1000, sc-81434) obtained from Santa Cruz; and anti-PHGDH (1:750, 13428), anti-CCR2 (1:1000, 12199) obtained from CST; and anti-α-Tubulin (1:1000, 1224-1-AP) and secondary antibodies horseradish peroxidase (HRP)-conjugated anti-rabbit Ig (H + L) (1:5000, SA00001-2) and HRP-conjugated anti-mouse Ig(H + L) (1:5000, SA00001-1) were purchased from Proteintech.
Macrophage migration assayIn vitro migration assays were performed using tissue culture-treated cell inserts with 8.0 µM pore size PET membranes (cellQART). A549 or AE17 cancer cells were seeded at a density of 3 ×105 cells/well in a 24-well plate and incubated overnight. For some experiments, 100 nM rapamycin or 1 mM DM-αKG was added to the upper chamber. The media were then removed, and the cells were washed twice with PBS. Fresh medium containing 0.5% heat-inactivated FBS was added to the wells. Next, 0.5 × 106Phgdhfl/flCx3cr1-Cre or Phgdhfl/fl BMDMs were seeded in 200 μL DMEM supplemented with 0.5% heat-inactivated FBS on the top of the membrane chamber and allowed to incubate for 10 min before the chamber was submerged into the lower compartment of the 24-well plate. After 24 h, the insert was removed, washed and fixed in 4% paraformaldehyde (PFA) for 20 min. Nonmigrated cells on the upper side of the membrane were removed with cotton swabs, and the insert was then submerged in 0.2% crystal violet solution for 10 min for cell staining. The insert was dried overnight following washing with tap water. The migrated cells were photographed in 6 random fields and quantified manually by counting single cells under a Nikon ECLIPSE Ti2 microscope (Nikon).
Seahorse extracellular flux analysisThe macrophage OCR and ECAR were measured as previously described [55]. Briefly, Phgdhfl/flCx3cr1-Cre and Phgdhfl/fl BMDMs were plated in 6-well plates at a density of 1 × 106 cells per well. After incubating with TCM for 24 h, the cells were scraped and transferred to Cell-Tak (Corning)-precoated XF24e-cell culture plates for immediate adhesion with XF assay medium (Agilent Technologies). Before the measurements were taken, the cells were placed in a CO2-free incubator at 37 °C for 1 hour. In the meantime, a utility plate containing 1 mL of calibrant solution was prepared to hydrate the XF extracellular flux cartridge. The utility plate was incubated overnight in a CO2-free incubator at 37 °C. Following the instructions of the glycolysis stress assay from Agilent, glucose (30 mM), oligomycin (30 μM) and 2-deoxy-D-glucose (2-DG, 50 mM) were added to each well. Real-time extracellular acidification (ECAR) was measured using a seahorse analyzer. For the mitochondrial stress assay, the cells were incubated in XF assay medium supplemented with 2 mM glutamine, 1 mM pyruvate and 25 mM glucose. The final concentrations of oligomycin (3 μM), FCCP (3 μM), and a mixture of rotenone/antimycin A (0.5 μM) were injected into ports A, B, and C, respectively. The data were monitored for the ECAR and OCR, and the values were normalized to the cell number.
RNA-seq and bioinformatics analysisPhgdhfl/flCx3cr1-Cre and Phgdhfl/fl BMDMs were seeded in a 6-well plate at a density of 1 ×106 cells per well. After incubating with AE17-TCM for 24 h, total RNA was isolated using TRIzol reagent according to the manufacturer’s instructions. Library preparation was carried out at the Next Generation Sequencing (NGS) unit of the Vienna Biocenter Core Facility (VBCF, Vienna, Austria). The NEBNext Ultra Directional RNA Library Prep Kit (New England Biolabs) was used for preparing the sequencing libraries. The average length of the pooled libraries was 330–360 bp, and the libraries were sequenced on a NextSeq550 instrument (Illumina) using the 1 ×75 bp sequencing mode. The RNA-Seq reads were mapped to the reference mouse mm10 genome using TopHat2 [56], and fragments per kilobase of transcript per million fragments mapped (FPKM) were calculated via HT-seq [57]. The differential gene expression analysis was conducted by DESeq2 [58] with the following statistical cutoffs: genes with low counts greater than 10, adjusted p value less than 0.05, and an absolute log2-fold change greater than 1.5.
siRNA transfectionPsat1 siRNA was obtained from Santa Cruz, and siRNA transfection was performed as described earlier [55]. Briefly, RAW 264.7 macrophages were seeded onto a 6-well plate at a density of 0.6 × 106 cells/well. Once the cells reached 70–80% confluence, they were transfected with either control siRNA or Psat1 siRNA using Lipofectamine 3000 Transfection Reagent (Invitrogen) according to the manufacturer’s instructions. After 48 h of transfection, the cells were gently washed with PBS and then incubated in AE17-TCM supplemented with or without DM-αKG for 24 h. The cells were subsequently harvested for analysis of mRNA expression and metabolites.
Pulsed stable isotope-resolved metabolomicsStable isotope tracing experiments were performed as described previously with some modifications [59]. Briefly, differentiated BMDMs were seeded at a density of 1 ×106 cells per well in DMEM and incubated overnight. The medium was then replaced with AE17-TCM, and the cells were incubated for 24 h. Two hours prior to harvest, a second medium was used to expose the cells to DMEM without glucose or serine (D9802-01, USBiological) supplemented with 4.5 g/L U-[13C]-glucose (Cambridge Isotope Laboratories; CLM-1396), 0.4 mM U-[13C]-serine (Cambridge Isotope Laboratories; CLM-1574-H) or 1 mM U-[13C]-glutamine (Cambridge Isotope Laboratories; CLM-1822-H). The cells were washed twice with HEPES buffer (5 mM HEPES, 100 mM NaCl, pH 7.4) and quenched by adding 1 mL of 50% precooled methanol (−80 °C) containing 2.5 nM phenyl β-D-glucopyranoside (Sigma) as an internal control. Cell lysates were collected in polypropylene tubes by scraping, followed by the addition of 200 μL of chloroform. The samples were shaken for 1 hour at 4 °C. After centrifugation, the supernatant was transferred to a new tube and dried in a SpeedVac (Labogene). Next, 15 μL of methoxyamine hydrochloride solution (40 mg/mL in pyridine) was added to the dried fraction, and the mixture was then incubated for 90 min at 30 °C. Subsequently, 60 μL of N-methyl-N-trimethylsilyltrifluoroacetamide (MSTFA) was added, and the mixture was incubated for 30 min at 37 °C. The reaction mixture was centrifuged for 10 min at 4 °C and 21,000 × g, after which the supernatants were transferred to glass vials with microinserts. The metabolites were measured using GC‒MS following standard protocols [60]. Data processing and natural 13C labeling correction were performed using the Data Extraction for Stable Isotope-labeled metabolites (DExSI) software [61]. The default settings were used except for the following parameters. The number of points on either side of the apex and scan window was set to 10. Mass isotopomer fraction labeling was determined by integrating metabolite ion fragments (Supplementary Table S3).
Metabolomic analysis by GC‒MS and LC‒MSCellular metabolites were extracted and analyzed according to previously established methods with modifications [53]. Briefly, Phgdhfl/flCx3cr1-Cre and Phgdhfl/fl BMDMs were seeded at a density of 1 × 106 cells/well in DMEM and incubated overnight. The medium was then replaced with AE17-TCM for 24 h. The cells were washed twice with 0.9% NaCl and quenched by adding 80% ice-cold methanol containing 2.5 nM phenyl β-D-glucopyranoside (GC‒MS) or 10 nM ampicillin (LC‒MS) as the internal extraction standard. The extraction samples were incubated for 15 min at 4 °C and then centrifuged for 10 min at 21,000 × g. The supernatant was transferred to a fresh polypropylene tube and dried in a SpeedVac. The cell pellets were lysed in RIPA buffer, after which the protein levels were measured for normalization purposes. For measurements of αKG, serine and glycine levels in the blood of Phgdhfl/flCx3cr1-Cre and Phgdhfl/fl tumor-bearing mice, blood was drawn weekly from the facial vein and incubated for 2 h at 4 °C. Then, 10 μL of serum was collected after centrifugation, and metabolites were extracted in 1 mL of precooled methanol. The extraction samples were incubated for 15 min at 4 °C, and then centrifuged for 10 min at 21,000 × g. The supernatants were subsequently dried in a SpeedVac system. For GC‒MS analysis, sample derivatization was carried out as described above. The injection volume of each sample was 1 μL, and the samples were injected at a 1:5 split ratio. The total ion chromatogram was deconvoluted, and peak alignment and integration were performed using the software MS-DIAL [62].
LC‒MS analysis was performed using an UltiMate 3000 UHPLC System (Thermo Scientific) coupled to an LTQ-Orbitrap Elite mass spectrometer (Thermo Scientific). The extracted samples were dissolved in MS buffer (10 mM NH4OAc in mqH2O and 2% methanol, pH 6.9) and then centrifuged for 10 min at 21,000 × g. The supernatant was subsequently transferred to HPLC vials. The sample was separated using a Thermo ScientificTM Accucore™ Vanquish C-18+ (100 × 2.1 mm, 1.5 µm particle size) UHPLC column equipped with a Thermo ScientificTM Accucore™ Defender C18 guard cartridge (10 × 2.1 mm, 2.6 µm particle size). The sample injection volume was 5 µL. The mobile phase system consisted of a mixture of solvent A (10 mM NH4OAc in mqH2O, pH 6.9) and solvent B (LC‒MS grade methanol). A gradient elution method was used for the analysis: 0–1 min 5% B, 5–30 min linear gradient to 85% B, 0.1 min back to 0% B, 10 min 0% B. The flowrate was set to 0.25 mL/min, and the column oven temperature was 30 °C. MS analysis was performed with the following ion source parameters: spray voltage, 4 kV; capillary temperature, 350 °C; sheath gas, 35; and auxiliary gas, 10. Mass spectrometry was performed in positive ion mode. The full MS scan range was 100–1200 m/z with a resolution of 120,000. The normalized collision energy for collision-induced dissociation (CID) was set to 35%. Xcalibur software (Thermo Scientific) and MS-DIAL were used for data analysis and interpretation.
Statistical analysisAll of the data are presented as mean ± SEM unless otherwise noted. All of the statistical analyses were performed using Prism v9 (GraphPad Software) or Excel (Microsoft). Statistical significance was analyzed using an unpaired Student’s t test or two-way ANOVA with Tukey’s multiple comparisons test. Differences with p < 0.05 were considered to be statistically significant.
Comments (0)