Patients and tissue specimen collection
The research protocol was conducted in alignment with the principles of the Declaration of Helsinki. Prior to the commencement of the study, written consents were secured from all participating patients. Tissue samples, both malignant and adjacent normal, were procured from patients with bladder cancer (BCa) undergoing surgical procedures. Comprehensive follow-ups were conducted for all patients, with overall survival (OS) being calculated from the surgery date to either the date of demise or the last recorded follow-up for those still alive.
Cell culture and transfection
In our research, we utilized a collection of BCa cell lines, namely T24 and BIU-87, with SVHUC-1 cells serving as the standard control. These cell lines were sourced from the American Type Culture Collection (ATCC, Manassas, VA, USA) and the National Infrastructure of Cell Line Resource in China. Standard culture conditions for the BCa cell lines involved Roswell Park Memorial Institute (RPMI) 1640 or Dulbecco’s Modified Eagle Medium (DMEM) mediums enriched with 10% fetal bovine serum (sourced from Invitrogen, Carlsbad, CA, USA). These cells were incubated at 37 °C in an environment with 5% CO2. For transfection procedures, we employed lipo3000 (from Invitrogen) in adherence to the guidelines provided by the manufacturer, ensuring a consistent plasmid quantity of 1 μg for our experimental protocols.
To develop the cisplatin-resistant bladder cancer cell lines, we employed a gradual adaptation approach. Initially, the T24 and 5637 bladder cancer cell lines were exposed to a sub-lethal concentration of cisplatin. This initial dose was carefully chosen to be just below the level that induces significant cell death, ensuring the survival of a small population of less sensitive cells. Over the course of 6 months, we incrementally increased the cisplatin concentration after each cell adaptation and recovery phase. This method allowed for the selection and proliferation of cells with heightened resistance to cisplatin.
circRNA microarray
From the Cancer Center, we procured samples comprising four BCa specimens and their corresponding adjacent normal tissues. The protocols outlined by Arraystar (Rockville, MD, USA) guided our sample preparation and microarray hybridization processes. To isolate circular RNAs, linear RNAs were eliminated using RNase R digestion (sourced from Epicentre Technologies, Madison, WI, USA). These circRNAs were then transcribed into fluorescent circRNA using the Arraystar Super RNA Labeling Kit. The fluorescently labeled circRNAs were subsequently hybridized to the Arraystar Human circRNA Array V2 (8 × 15 K, Arraystar) and the results were captured using the Agilent Scanner G2505C (Jamul, CA, USA). To identify circRNAs with significant differential expression (with an |average normalized fold change| of ≥ 1.3) between the groups, we employed fold change thresholds.
RNA quantitative real-time polymerase chain reaction
RNA was isolated utilizing the TRIzol reagent (sourced from Invitrogen, Carlsbad, USA). From the extracted RNA, cDNA was synthesized using two micrograms as the starting material. Subsequently, 1 µl of this cDNA (equivalent to 0.2 µg) was employed for polymerase chain reaction (PCR) amplification. The real-time polymerase chain reaction (RT-PCR) was executed using SYBR Green SuperMix (obtained from Roche, Basel, Switzerland) and the ABI7900HT Fast Real-Time PCR system (from Applied Biosystems, CA, USA), with 1 µl cDNA serving as the template. For normalization, β-actin or U3 was utilized as an internal reference.
Actinomycin D and RNase R treatment
BCa cells were seeded into six-well plates and, after 24 h, reached approximately 60% confluency. Subsequently, the cells were exposed to either 5 μg/ml actinomycin D or dimethyl sulfoxide (DMSO) and harvested at specified intervals. For RNA processing, 2 μg of total RNA was treated with 3 U/μg of RNase R (sourced from Epicentre Technologies, Madison, WI, USA) and incubated for 15 min at 37 °C. Post actinomycin D or RNase R treatment, the expression levels of circ_104797 and associated mRNAs were assessed using quantitative RT-PCR (qRT-PCR).
Nuclear and cytoplasmic extraction
Cytoplasmic and nuclear components were separated using the PARIS™ Kit (AM1556, Thermo Fisher Scientific, Waltham, USA) as per the manufacturer’s guidelines. In brief, BCa cells were subjected to lysis in cell fraction buffer and incubated on ice for 10 min. Post incubation, the mixture was centrifuged at 500 × g for 3 min at 4 °C and the resulting supernatant, representing the cytoplasmic fraction, was carefully collected. The remaining pellet was rinsed with cell fraction buffer to obtain the nuclear fraction.
RNA fluorescence in situ hybridization (FISH)
The probe sequence specific to circ_104797 was procured from Sangon Biotech (Shanghai, China). Cells, once fixed, were rinsed with phosphate-buffered saline (PBS) and subsequently treated with RNase R at 37 °C for a duration of 15 min, followed by a second fixation. The cell mixture was then dispensed onto sterilized glass slides and dehydrated sequentially using 70%, 80%, and 100% ethanol. Hybridization ensued in a humidified, dark chamber at 37 °C overnight. Post-hybridization, the slides were rinsed twice with a solution of 50% formamide/2 × SSC for 5 min each. Subsequently, the slides were treated with reagents from the Alexa FluorTM 488 Tyramide SuperBoost™ Kit (Thermo Fisher Scientific, Waltham, USA) for 30 min and then sealed using parafilm embedded with 4′,6-diamidino-2-phenylindole (DAPI). Fluorescent images were captured using a fluorescence microscope. ImageJ software was employed for fluorescence intensity analysis, while the OLYMPUS FV1000 software was utilized to assess Pearson’s correlation coefficient.
Methylated RNA immunoprecipitation PCR (MeRIP-qPCR)
To quantitatively assess m6A-modified mRNA, MeRIP-qPCR was employed. Post mRNA extraction, the anti-m6A antibody and anti-IgG (sourced from Cell Signaling Technology) were bound to protein A/G magnetic beads in immunoprecipitation (IP) buffer, which contained 140 mM NaCl, 20 mM Tris (pH 7.5), 2 mM EDTA, and 1% NP-40 along with RNase and protease inhibitors. This mixture was incubated overnight at 4 °C. Subsequent to this incubation, the RNA-bead complex was separated using elution buffer. The final step involved determining RNA enrichment via qRT-PCR, where the enrichment was calculated using the 2-ΔΔCt method, comparing the eluate to the input sample.
RNA-binding protein immunoprecipitation (RIP)
The RIP assay was conducted using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore) as per the manufacturer’s guidelines. In essence, BCa cells, post 48-h transfection, were collected and subjected to lysis in RIP lysis buffer, with incubation on ice for 30 min. Following centrifugation, the resultant supernatant was combined with 30 μl of Protein-A/G agarose beads (sourced from Roche, USA) and specific antibodies. This mixture was incubated overnight. Subsequently, the immune complexes underwent centrifugation and were washed six times using a washing buffer. Protein analysis from the bead-bound complexes was carried out via western blotting, while the RNA extracted from the immunoprecipitation was evaluated using qRT-PCR.
RNA pull-down
Using streptavidin-coated magnetic beads (Invitrogen, Carlsbad, USA), the biotinylated RNA complex was isolated from cell lysates as per the manufacturer’s guidelines. The enrichment of circRNA in the isolated fractions was determined using qRT-PCR. Proteins bound to the beads were subsequently eluted and subjected to sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Western blotting was employed to identify and analyze the proteins present in the captured complex.
Luciferase reporter assay
The circRNA sequence was integrated into the p-GLO Dual-Luciferase vector (Vigenebio, Maryland, USA), with specific mutations introduced at the binding sites. BCa cells, seeded in 24-well plates at 30% confluency, were co-transfected 24 h later with 800 ng of the p-GLO Dual-Luciferase reporter and 800 ng of vectors. After an incubation period of 48 h, the dual luciferase reporter assay system (Promega, Madison, WI) was employed to determine the relative luciferase activity, which was expressed as the Firefly to Renilla luciferase activity ratio. The Renilla luciferase activity served as an internal normalization control. For the ALKBH5 overexpression samples, the Luc/Rluc ratio was further standardized to the control group’s value.
Cell proliferation assay
Cellular proliferation was evaluated using the CCK-8 assay kit (Transgen, China). Cells were plated in 96-well plates at a concentration of 3 × 103 cells per well. Post-seeding, cells were incubated for specific durations:12, 24, 48, and 72 h. After each interval, CCK-8 reagent was added to the wells, and the plates were incubated at 37 °C for an additional 3 h. The optical density of each well was subsequently recorded at 450 nm using a microplate spectrophotometer.
Wound healing migration assays
Wound healing assays were performed using a standard protocol. Cells were seeded in six-well chamber slides at a concentration of 5 × 105 cells/well. A sterile 10 μl pipette tip was employed to create a linear scratch in the cell monolayer. After a 48-h incubation period, images were taken to assess the migration of cells into the wound area. The percentage of wound closure was calculated using the formula: wound closure (%) = [(initial wound width − width after 48 h)/initial wound width] × 100%.
Cell apoptosis assay
In a 12-well plate, cells transfected with the designated vector were seeded at a concentration of 2 × 105 cells per well, ensuring 70–80% confluency. Following a 48-h incubation, apoptosis was evaluated using the caspase-3 ELISA assay kit (Hcusabio, China). Given its role in mediating DNA and cytoskeletal protein degradation, caspase-3 is a critical marker for apoptotic pathways and inflammatory responses. Each experimental setup was performed in triplicate to ensure consistency and precision.
Xenograft model
In this research, we incorporated female BALB/c nude mice aged between 4 and 5 weeks. These mice were divided into two distinct groups, each consisting of six individuals. The living conditions for these mice were maintained at specific-pathogen-free standards in line with conventional animal housing protocols. We administered a subcutaneous injection of 5 million transfected bladder cancer cells into the right flank region of each mouse. To track the progression of the tumors, we measured their volumes on a weekly basis. At 4 weeks following the injection, the mice were humanely euthanized under anesthesia, and the tumors were extracted from the subcutaneous tissues. These excised tumors were then analyzed for both their volume and weight. Moreover, our methods for handling and experimenting with the animals were in strict compliance with the ethical guidelines for animal research as set by our institution.
Bioinformatic analysis
To investigate the potential biological functions of circ_104797, we performed a prediction by SRAMP (http://www.cuilab.cn/sramp) and RMBase v2.0. CircRNA-miRNA interactions were predicted using Arraystar’s miRNA target prediction software based on TargetScan and miRanda.
Statistical analysis
Data are expressed as mean ± standard deviation (SD). Statistical evaluations were conducted using GraphPad Prism 8.0 (GraphPad, San Diego, CA, USA) and SPSS version 20.0 (IBM, SPSS, Chicago, IL, USA). For pairwise comparisons, either a two-sided Student’s t-test or a two-tailed Mann–Whitney U-test was applied, contingent on the data distribution. For multiple group comparisons, one-way analysis of variance (ANOVA) was employed, followed by Bonferroni post hoc tests for further analysis. A P-value less than 0.05 was deemed statistically significant. In figures or accompanying legends, significance levels are indicated as *P < 0.05 and **P < 0.01.



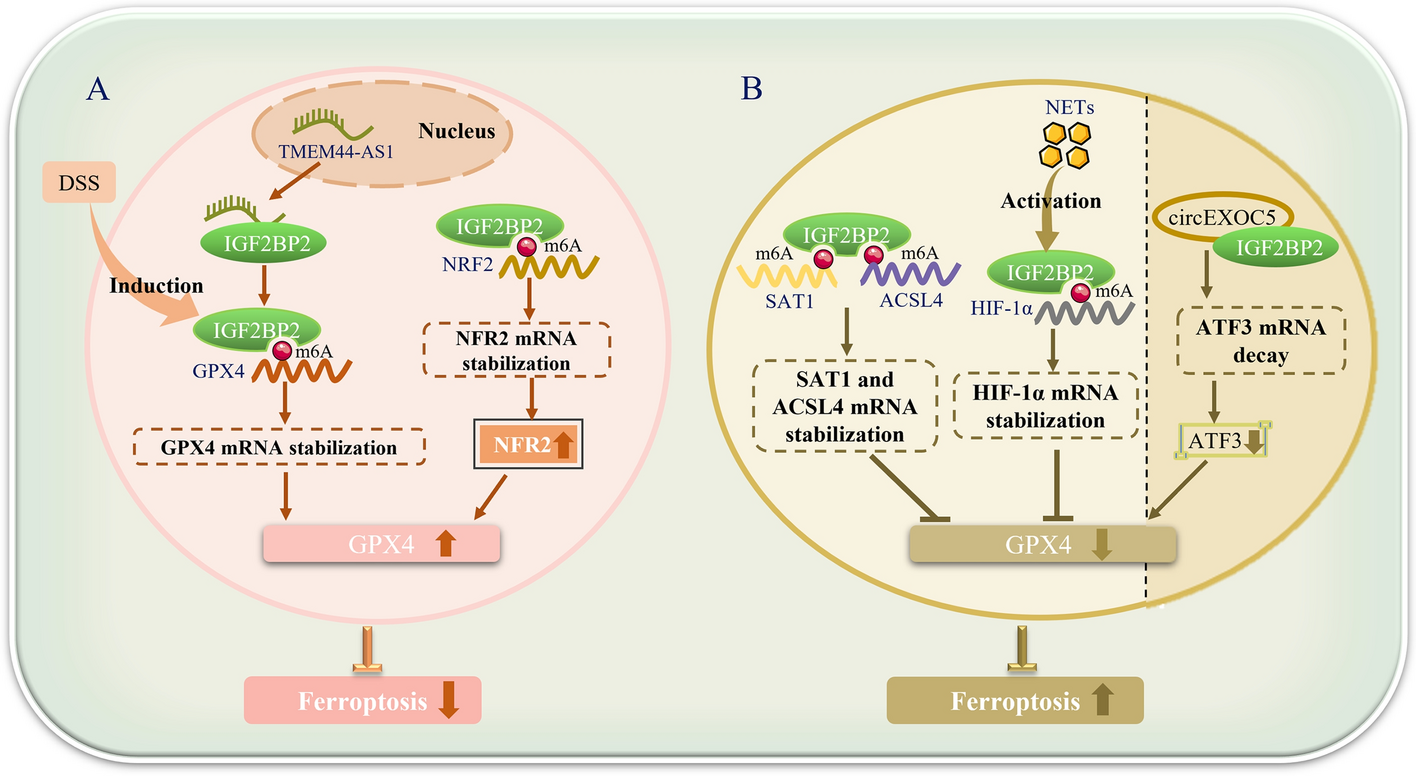

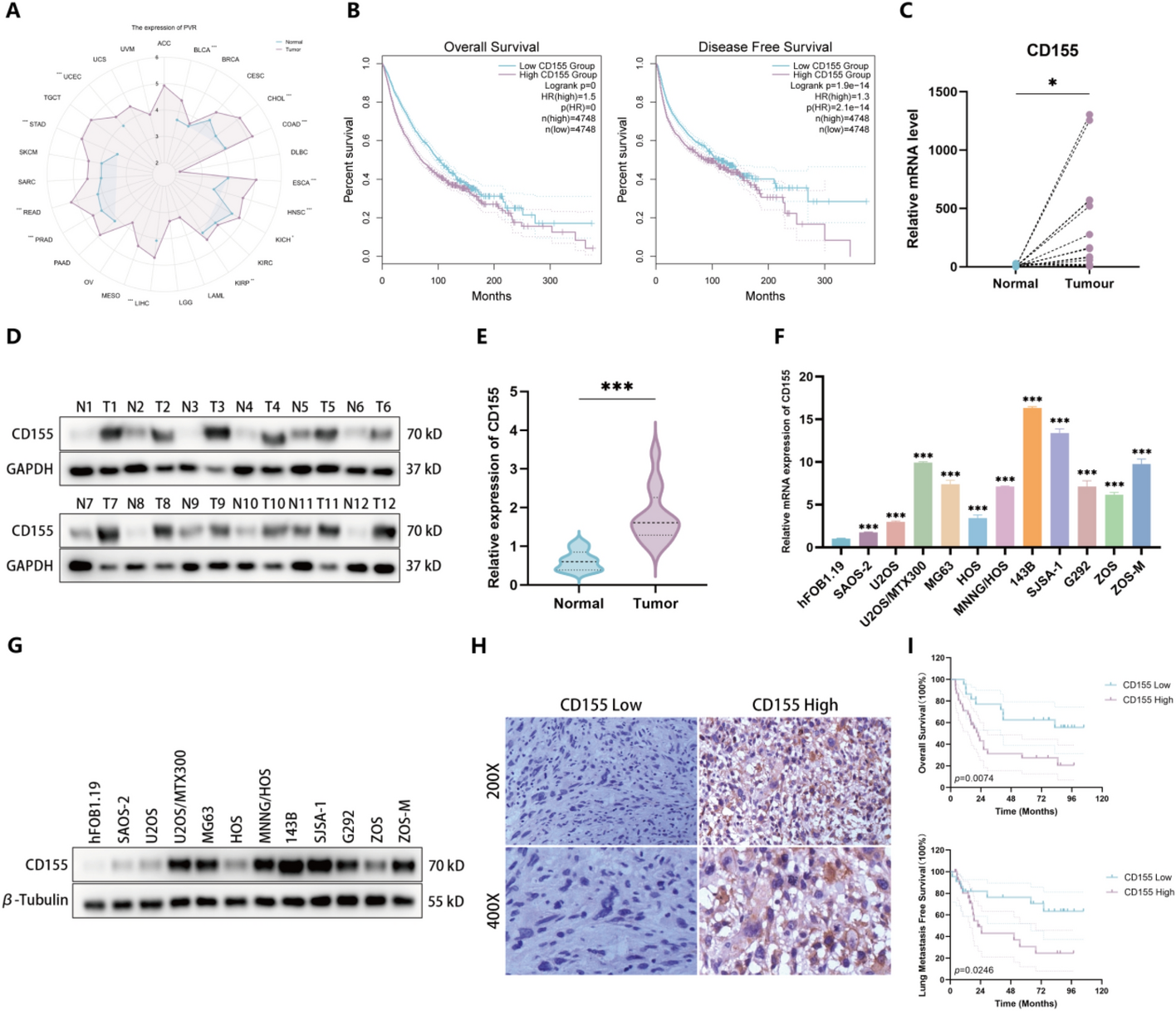

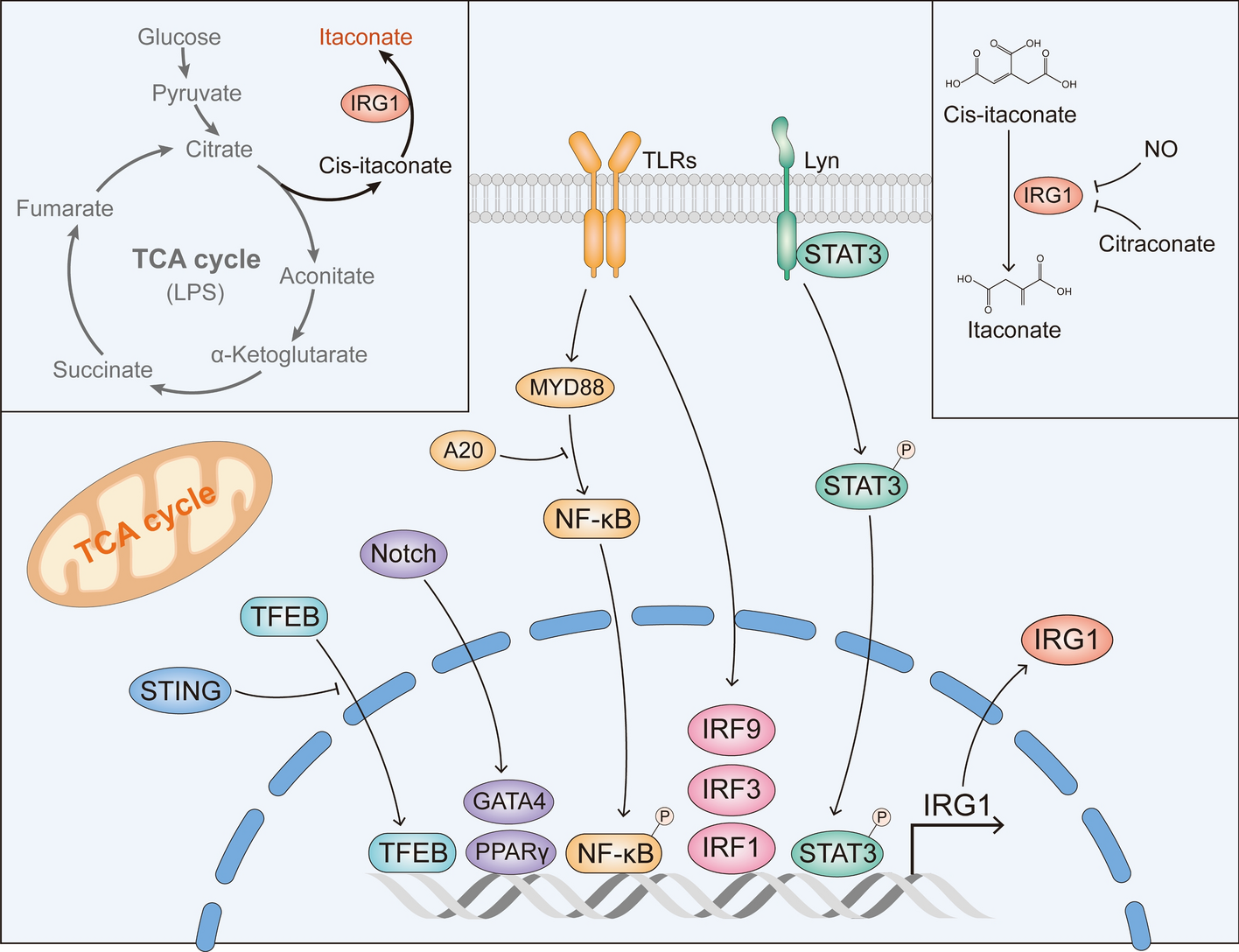


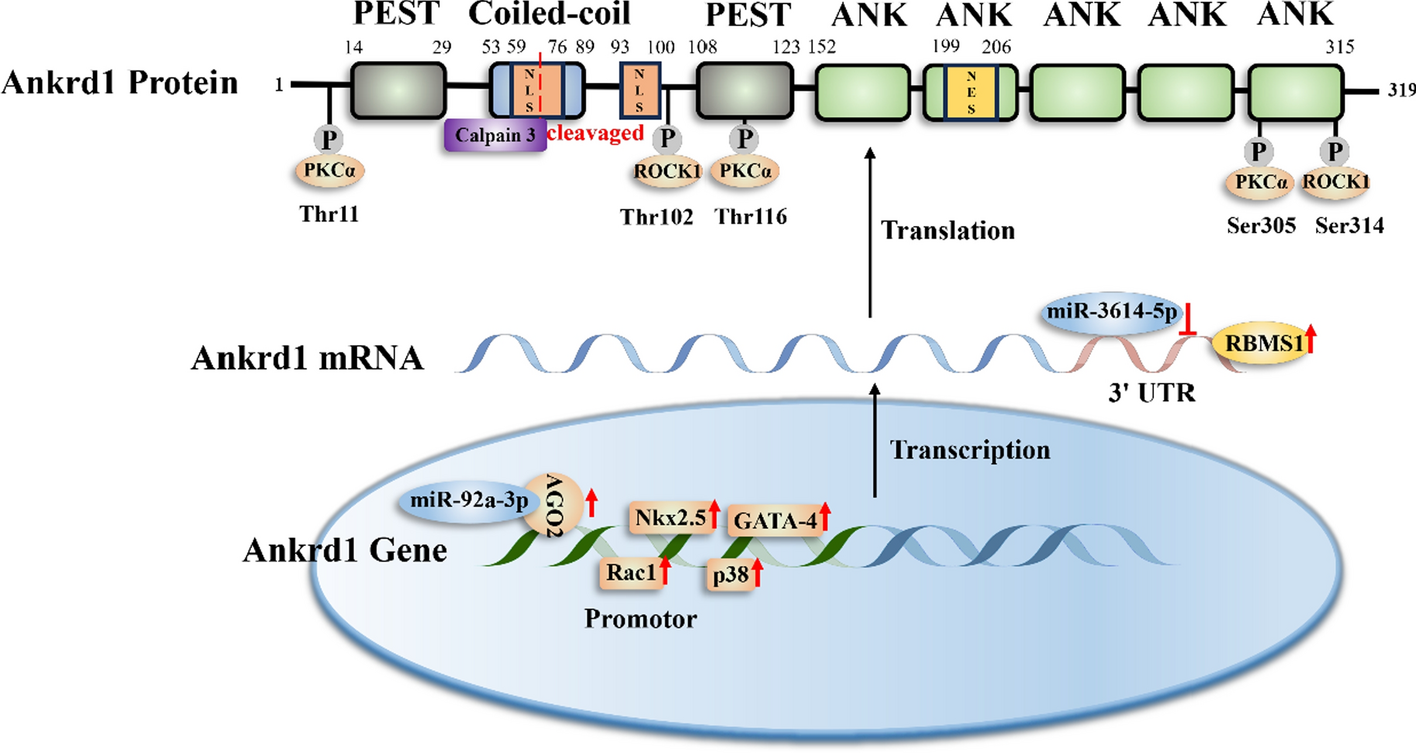
Comments (0)